原文地址:https://www.jianshu.com/p/1174a53abe7d
作业要求
需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量!
作业,理解测序reads,GC含量,质量值,接头,index,fastqc的全部报告,搜索中文教程,并发在论坛上面。
来源于生信技能树:http://www.biotrainee.com/forum.php?mod=viewthread&tid=1750#lastpost
实验步骤
1. 将 sra 数据转化成 fastq 格式(先把所有的sra放到一个文件夹里,姑且命名为SRR,然后 cd ~/SRR进入这个文件夹)
for i in {56..62}
do
fastq-dump --gzip --split-3-O /Users/chengkai/Desktop/zhuanlu_files -A SRR35899${i}.sra
done
--gzip 压缩格式为gzip
--split-3 如果是双端测序输出两个文件,如果不是只输出一个文件
-0 输出文件路径
“/Users/chengkai/Desktop/zhuanlu_files” 这里改成你自己的文件路径
-A 输入文件路径
搞定之后,会生成两个文件,列举其中一个 SRR3589956.sra_1.fastq.gz
然后所有的文件再放入另外一个文件夹(姑且命名SRA),然后在cd ~/SRA进入这个文件夹里面
for i in `seq 56 62`
> do
> fastqc SRR35899${i}.sra_1.fastq.gz
> done
或者 fastqc SRR35899${i}.sra_1.fastq.gz

质量解读
html 格式用浏览器打开
基本信息
Enconding: 测序平台版本号
Total Sequence: reads 的数量
Sequence length: 总的序列数
%GC GC比,这个指标有物种意义,用于区别物种,一般人类42%

image.png
每个read各位置碱基的测序质量
横轴碱基的位置(1-51),纵轴是质量分数,20表示1%的错误率,30表示0.1%
红色线代表中位数,蓝色代表平均数,黄色是25%-75%区间,触须是10%-90%区间
Warning 报警 如果任何碱基质量低于10,或者是任何中位数低于25
Failure 报错 如果任何碱基质量低于5,或者是任何中位数低于20
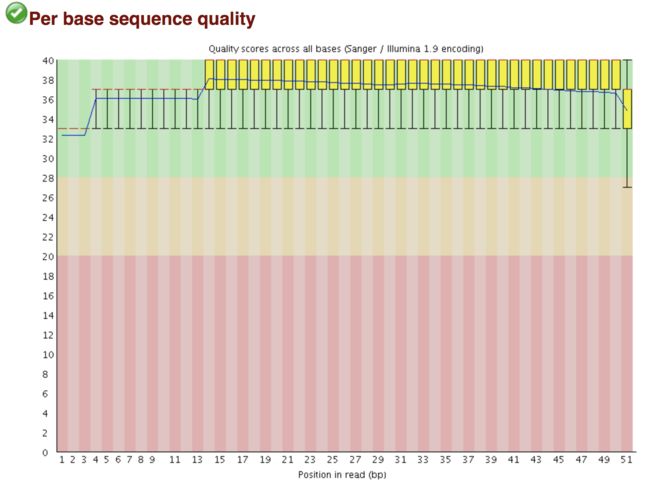
image.png
偏离度
横轴碱基的位置(1-51)
纵轴是tail的Index编号
检查reads中每一个碱基位置在不同的测序小孔之间的偏离度,蓝色代表偏离度小,质量好,越红代表偏离度越大,质量越差。
这个图主要是为了防止,在测序过程中,某些tail受到不可控因素的影响而出现测序质量偏低
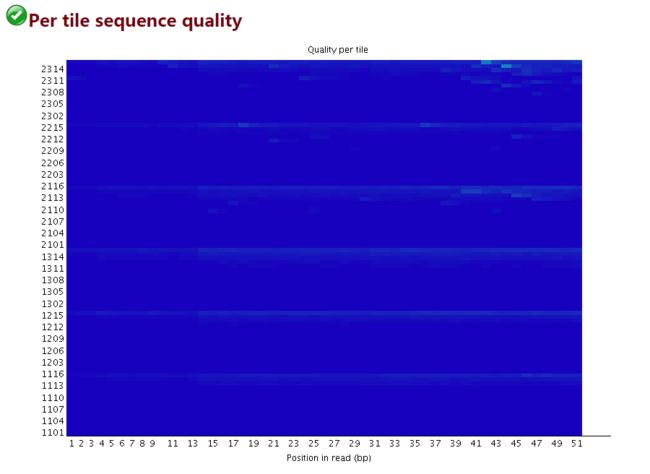
image.png
reads质量的分布
横轴表示Q值,0-40
纵轴是每个值对应的reads数目
当峰值小于27时,警告;当峰值小于20时,fail。我的报告峰值在38

image.png
GC 含量统计
横轴碱基的位置(1-51)
纵轴是碱基含量百分比
图中四条线代表A T C G在每个位置平均含量
当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。
本结果前10个位置,每种碱基频率有明显的差别,说明有污染。
当任一位置的A/T比例与G/C比例相差超过10%,报"WARN";当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"
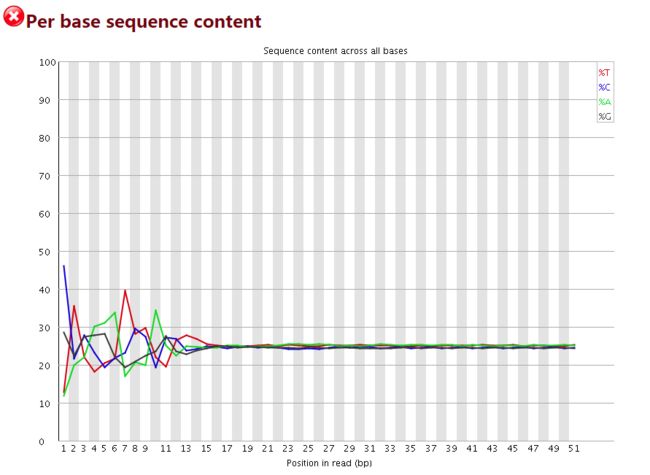
image.png
序列平均GC含量分布图
横轴是百分比; 纵轴是每条序列GC含量对应的数量
蓝色的线是程序根据经验分布给出的理论值,红色是真实值,两个应该比较接近才比较好
当红色的线出现双峰,基本肯定是混入了其他物种的DNA序列
偏离理论分布的reads超过15%时,报"WARN";偏离理论分布的reads超过30%时,报"FAIL"
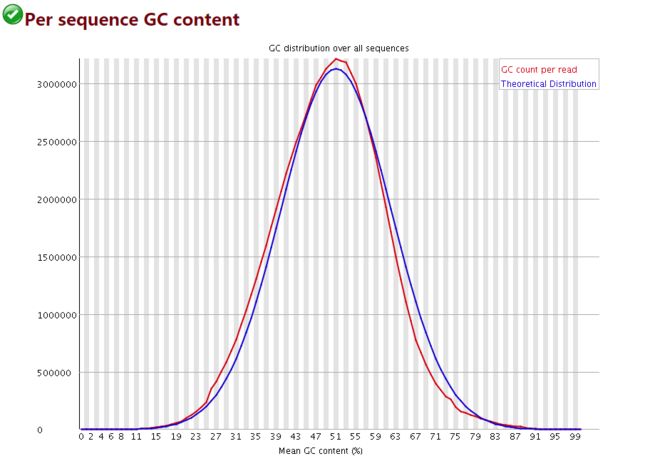
image.png
各位置N的reads比率
当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”,统计N的比率
正常情况下,N值非常小
当任意位置的N的比例超过5%,报"WARN";当任意位置的N的比例超过20%,报"FAIL"

image.png
reads 长度分布
每次测序仪测出来的长度在理论上应该是完全相等的
当reads长度不一致时报"WARN";当有长度为0的read时报“FAIL”
当测序的长度不同时,如果很严重,则表明测序仪在此次测序过程中产生的数据不可信

image.png
统计不同拷贝数的reads的频率
横坐标是duplication的次数,纵坐标是duplicated reads的数目,以unique reads的总数作为100%
测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在
当非unique的reads占总数的比例大于20%时,报"WARN";当非unique的reads占总数的比例大于50%时,报"FAIL"

image.png

image.png
接头含量
此图衡量的是序列中两端adapter的情况
如果在当时fastqc分析的时候-a选项没有内容,则默认使用图例中的四种通用adapter序列进行统计
本例中adapter都已经去除,如果有adapter序列没有去除干净的情况,在后续分析的时候需要先使用cutadapt软件进行去接头

image.png
重复短序列
这个图统计的是,在序列中某些特征的短序列重复出现的次数
我们可以看到1-8bp的时候图例中的几种短序列都出现了非常多的次数,一般来说,出现这种情况,要么是adapter没有去除干净,而又没有使用-a参数;要么就是序列本身可能重复度比较高,如建库PCR的时候出现了bias
对于这种情况,我的办法是可以cut掉前面的一些长度,可以试着cut 1bp

image.png
参考文献
http://fbb84b26.wiz03.com/share/s/3XK4IC0cm4CL22pU-r1HPcQQ2irG2836uQYm2iZAyh1Zwf3_(青山屋主)
www.biotrainee.com/thread-2034-1-1.html(laofuzi)
http://www.jianshu.com/p/14fd4de54402(lxmic)
https://zhuanlan.zhihu.com/p/20731723(孟浩巍)