宏基因组数据分析:差异分析(LEfSe安装使用及LDA score计算)
文章目录
-
- 导读
- 原理
- LDA score计算
- 安装
- 分析
-
- 文件输入格式
- 数据格式转换
- lefse分析
- 绘制lefse结果图
- 绘制特征条形图
- 绘制系统发育树图
- 参考资料
导读
- LEfSe(Linear discriminant analysis Effect Size)通过将用于统计显着性的标准检验与编码生物一致性和效果相关性的检验相结合,确定最有可能解释类别之间差异的特征。
- LEfSe分析可以实现多个分组之间的比较,还进行分组比较的内部进行亚组比较分析,从而找到组间在丰度上有显著差异的物种(即biomaker)。
原理
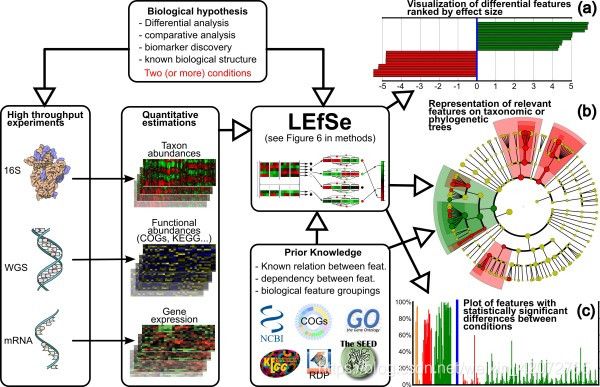
- 首先在多组样本中采用的非参数检验Kruskal-Wallis秩和检验检测不同分组间丰度差异显著的特征;
- 然后在上一步中获得的显著差异特征,用成组的Wilcoxon秩和检验进行组间差异分析(若没有亚组,该步跳过);
- 最后用线性判别分析(LDA)对数据进行分类和评估差异显著的物种的影响力(即LDA score)。
LDA score计算
计算步骤包括(详细计算过程看源码):
- 拟合lda模型,获取第一特征向量;
- 对第一特征向量进行标准化;
- 根据标准化后的第一特征向量,计算样本新坐标;
- 根据分组信息,计算组间距离,作为效应系数;
- LDA score = 效应系数 * 标准化后特征向量 ;
- 对LDA score进行log转换。
def test_lda_r(cls,feats,cl_sl,boots,fract_sample,lda_th,tol_min,nlogs):
fk = list(feats.keys())
means = dict([(k,[]) for k in feats.keys()])
feats['class'] = list(cls['class'])
clss = list(set(feats['class']))
for uu,k in enumerate(fk):
if k == 'class':
continue
ff = [(feats['class'][i],v) for i,v in enumerate(feats[k])]
for c in clss:
if len(set([float(v[1]) for v in ff if v[0] == c])) > max(float(feats['class'].count(c))*0.5,4):
continue
for i,v in enumerate(feats[k]):
if feats['class'][i] == c:
feats[k][i] = math.fabs(feats[k][i] + lrand.normalvariate(0.0,max(feats[k][i]*0.05,0.01)))
rdict = {
}
for a,b in feats.items():
if a == 'class' or a == 'subclass' or a == 'subject':
rdict[a] = robjects.StrVector(b)
else:
rdict[a] = robjects.FloatVector(b)
robjects.globalenv["d"] = robjects.DataFrame(rdict)
lfk = len(feats[fk[0]])
rfk = int(float(len(feats[fk[0]]))*fract_sample)
f = "class ~ "+fk[0]
for k in fk[1:]:
f += " + " + k.strip()
ncl = len(set(cls['class']))
min_cl = int(float(min([cls['class'].count(c) for c in set(cls['class'])]))*fract_sample*fract_sample*0.5)
min_cl = max(min_cl,1)
pairs = [(a,b) for a in set(cls['class']) for b in set(cls['class']) if a > b]
for k in fk:
for i in range(boots):
means[k].append([])
for i in range(boots):
for rtmp in range(1000):
rand_s = [lrand.randint(0,lfk-1) for v in range(rfk)]
if not contast_within_classes_or_few_per_class(feats,rand_s,min_cl,ncl):
break
rand_s = [r+1 for r in rand_s]
means[k][i] = []
for p in pairs:
robjects.globalenv["rand_s"] = robjects.IntVector(rand_s)
robjects.globalenv["sub_d"] = robjects.r('d[rand_s,]')
z = robjects.r('z <- suppressWarnings(lda(as.formula('+f+'),data=sub_d,tol='+str(tol_min)+'))')
robjects.r('w <- z$scaling[,1]')
robjects.r('w.unit <- w/sqrt(sum(w^2))')
robjects.r('ss <- sub_d[,-match("class",colnames(sub_d))]')
if 'subclass' in feats:
robjects.r('ss <- ss[,-match("subclass",colnames(ss))]')
if 'subject' in feats:
robjects.r('ss <- ss[,-match("subject",colnames(ss))]')
robjects.r('xy.matrix <- as.matrix(ss)')
robjects.r('LD <- xy.matrix%*%w.unit')
robjects.r('effect.size <- abs(mean(LD[sub_d[,"class"]=="'+p[0]+'"]) - mean(LD[sub_d[,"class"]=="'+p[1]+'"]))')
scal = robjects.r('wfinal <- w.unit * effect.size')
rres = robjects.r('mm <- z$means')
rowns = list(rres.rownames)
lenc = len(list(rres.colnames))
coeff = [abs(float(v)) if not math.isnan(float(v)) else 0.0 for v in scal]
res = dict([(pp,[float(ff) for ff in rres.rx(pp,True)] if pp in rowns else [0.0]*lenc ) for pp in [p[0],p[1]]])
for j,k in enumerate(fk):
gm = abs(res[p[0]][j] - res[p[1]][j])
means[k][i].append((gm+coeff[j])*0.5)
res = {
}
for k in fk:
m = max([numpy.mean([means[k][kk][p] for kk in range(boots)]) for p in range(len(pairs))])
res[k] = math.copysign(1.0,m)*math.log(1.0+math.fabs(m),10)
return res,dict([(k,x) for k,x in res.items() if math.fabs(x) > lda_th])
安装
# 新建虚拟环境,lefse依赖于python2
conda create -n lefse python=2.7
# 激活虚拟环境
source activate lefse
# conda安装lefse
conda install lefse
# 退出虚拟环境
source deactivate
报错:如果是r2py的问题,基本都是python版本的问题。
解决:虚拟环境中重新安装python2.7(推荐)。另一种方式是对虚拟环境中的conda降级,之后再进行安装。
报错:如果是axis_bgcolor()的问题,那就是matplotlib版本问题。
解决:虚拟环境中重新安装metaplotlib 2.0版本,conda install matplotlib=2.0(推荐)。另一种方式是将lefse-plot_cladogam.py文件中的axis_bgcolor()修改为facecolor()。
分析
文件输入格式
文本制表符(单个Tab)分隔的输入文件由特征列表,分类向量和可选的亚组变量和样本ID向量组成。元数据位于前几行,特征矩阵随后。
bodysite mucosal mucosal mucosal mucosal mucosal non_mucosal non_mucosal non_mucosal non_mucosal non_mucosal
subsite oral gut oral oral gut skin nasal skin ear nasal
id 1023 1023 1672 1876 1672 159005010 1023 1023 1023 1672
Bacteria 0.99999 0.99999 0.999993 0.999989 0.999997 0.999927 0.999977 0.999987 0.999997 0.999993
Bacteria|Actinobacteria 0.311037 0.000864363 0.00446132 0.0312045 0.000773642 0.359354 0.761108 0.603002 0.95913 0.753688
Bacteria|Bacteroidetes 0.0689602 0.804293 0.00983343 0.0303561 0.859838 0.0195298 0.0212741 0.145729 0.0115617 0.0114511
Bacteria|Firmicutes 0.494223 0.173411 0.715345 0.813046 0.124552 0.177961 0.189178 0.188964 0.0226835 0.192665
Bacteria|Proteobacteria 0.0914284 0.0180378 0.265664 0.109549 0.00941215 0.430869 0.0225884 0.0532684 0.00512034 0.0365453
Bacteria|Firmicutes|Clostridia 0.090041 0.170246 0.00483188 0.0465328 0.122702 0.0402301 0.0460614 0.135201 0.0115835 0.0537381
数据格式转换
数据分析前需要对数据格式进行转换,原始数据可以为16S那种reads count,也可以是宏基因组相对丰度数据,但需要按照指定格式,行为分组和物种信息,列为样本。-c指定分组行;-s指定亚组行,若没有可以不指定;-u指定样本编号;-o指定归一化后范围,主要针对宏基因组数据,目的是对相对丰度进行放大。
lefse-format_input.py hmp_aerobiosis_small.txt hmp_aerobiosis_small.in -c 1 -s 2 -u 3 -o 1000000
详细参数:
usage: lefse-format_input.py [-h] [--output_table OUTPUT_TABLE] [-f {
c,r}]
[-c [1..n_feats]] [-s [1..n_feats]] [-o float]
[-u [1..n_feats]] [-m {
f,s}] [-n int]
[-biom_c BIOM_CLASS] [-biom_s BIOM_SUBCLASS]
INPUT_FILE OUTPUT_FILE
LEfSe formatting modules
positional arguments:
INPUT_FILE the input file, feature hierarchical level can be
specified with | or . and those symbols must not be
present for other reasons in the input file.
OUTPUT_FILE the output file containing the data for LEfSe
optional arguments:
-h, --help show this help message and exit
--output_table OUTPUT_TABLE
the formatted table in txt format
-f {
c,r} set whether the features are on rows (default) or on
columns
-c [1..n_feats] set which feature use as class (default 1)
-s [1..n_feats] set which feature use as subclass (default -1 meaning
no subclass)
-o float set the normalization value (default -1.0 meaning no
normalization)
-u [1..n_feats] set which feature use as subject (default -1 meaning
no subject)
-m {
f,s} set the policy to adopt with missin values: f removes
the features with missing values, s removes samples
with missing values (default f)
-n int set the minimum cardinality of each subclass
(subclasses with low cardinalities will be grouped
together, if the cardinality is still low, no pairwise
comparison will be performed with them)
-biom_c BIOM_CLASS For biom input files: Set which feature use as class
-biom_s BIOM_SUBCLASS
For biom input files: set which feature use as
subclass
lefse分析
转换格式后,可以进行lefse差异分析。需要指定输入和输出文件。还可以通过-a指定组间比较检验水准阈值,-w指定成组比较检验水准阈值,-l指定lda score阈值。除了可以选择lda,还可以选择svm进行分析。
run_lefse.py hmp_aerobiosis_small.in hmp_aerobiosis_small.res
详细参数:
usage: run_lefse.py [-h] [-o str] [-a float] [-w float] [-l float]
[--nlogs int] [--verbose int] [--wilc int] [-r str]
[--svm_norm int] [-b int] [-e int] [-c int] [-f float]
[-s {
0,1,2}] [--min_c int] [-t str] [-y {
0,1}]
INPUT_FILE OUTPUT_FILE
LEfSe 1.0
positional arguments:
INPUT_FILE the input file
OUTPUT_FILE the output file containing the data for the visualization
module
optional arguments:
-h, --help show this help message and exit
-o str set the file for exporting the result (only concise textual
form)
-a float set the alpha value for the Anova test (default 0.05)
-w float set the alpha value for the Wilcoxon test (default 0.05)
-l float set the threshold on the absolute value of the logarithmic
LDA score (default 2.0)
--nlogs int max log ingluence of LDA coeff
--verbose int verbose execution (default 0)
--wilc int wheter to perform the Wicoxon step (default 1)
-r str select LDA or SVM for effect size (default LDA)
--svm_norm int whether to normalize the data in [0,1] for SVM feature
waiting (default 1 strongly suggested)
-b int set the number of bootstrap iteration for LDA (default 30)
-e int set whether perform the wilcoxon test only among the
subclasses with the same name (default 0)
-c int set whether perform the wilcoxon test ing the Curtis's
approach [BETA VERSION] (default 0)
-f float set the subsampling fraction value for each bootstrap
iteration (default 0.66666)
-s {
0,1,2} set the multiple testing correction options. 0 no correction
(more strict, default), 1 correction for independent
comparisons, 2 correction for independent comparison
--min_c int minimum number of samples per subclass for performing
wilcoxon test (default 10)
-t str set the title of the analysis (default input file without
extension)
-y {
0,1} (for multiclass tasks) set whether the test is performed in
a one-against-one ( 1 - more strict!) or in a one-against-
all setting ( 0 - less strict) (default 0)
绘制lefse结果图
将lefse分析结果绘图。可以根据需求指定绘图格式。
lefse-plot_res.py hmp_aerobiosis_small.res hmp_aerobiosis_small.png
详细参数:
usage: lefse-plot_res.py [-h] [--feature_font_size FEATURE_FONT_SIZE]
[--format {
png,svg,pdf}] [--dpi DPI] [--title TITLE]
[--title_font_size TITLE_FONT_SIZE]
[--class_legend_font_size CLASS_LEGEND_FONT_SIZE]
[--width WIDTH] [--height HEIGHT] [--left_space LS]
[--right_space RS] [--orientation {
h,v}]
[--autoscale {
0,1}] [--background_color {
k,w}]
[--subclades N_SCL]
[--max_feature_len MAX_FEATURE_LEN]
[--all_feats ALL_FEATS] [--otu_only]
[--report_features]
INPUT_FILE OUTPUT_FILE
Plot results
positional arguments:
INPUT_FILE tab delimited input file
OUTPUT_FILE the file for the output image
optional arguments:
-h, --help show this help message and exit
--feature_font_size FEATURE_FONT_SIZE
the file for the output image
--format {
png,svg,pdf}
the format for the output file
--dpi DPI
--title TITLE
--title_font_size TITLE_FONT_SIZE
--class_legend_font_size CLASS_LEGEND_FONT_SIZE
--width WIDTH
--height HEIGHT only for vertical histograms
--left_space LS
--right_space RS
--orientation {
h,v}
--autoscale {
0,1}
--background_color {
k,w}
set the color of the background
--subclades N_SCL number of label levels to be dislayed (starting from
the leaves, -1 means all the levels, 1 is default )
--max_feature_len MAX_FEATURE_LEN
Maximum length of feature strings (def 60)
--all_feats ALL_FEATS
--otu_only Plot only species resolved OTUs (as opposed to all
levels)
--report_features Report important features to STDOUT
绘制特征条形图
另外,可以分别对指定的物种,或者所以差异物种绘制条形图。
# 单个特征
lefse-plot_features.py -f one --feature_name "Bacteria.Bacteroidetes" hmp_aerobiosis_small.in hmp_aerobiosis_small.res Bacteroidetes.png
# 差异特征
lefse-plot_features.py -f diff --archive zip hmp_aerobiosis_small.in hmp_aerobiosis_small.res biomarkers.zip
详细参数:
usage: lefse-plot_features.py [-h] [--width WIDTH] [--height HEIGHT]
[--top TOP] [--bot BOT]
[--title_font_size TITLE_FONT_SIZE]
[--class_font_size CLASS_FONT_SIZE]
[--class_label_pos {
up,down}]
[--subcl_mean {
y,n}] [--subcl_median {
y,n}]
[--font_size FONT_SIZE] [-n flt]
[--format {
png,pdf,svg}] [-f {
all,diff,one}]
[--feature_name FEATURE_NAME]
[--feature_num FEATURE_NUM]
[--archive {
zip,none}]
[--background_color {
k,w}] [--dpi DPI]
INPUT_FILE INPUT_FILE OUTPUT_FILE
Cladoplot
positional arguments:
INPUT_FILE dataset files
INPUT_FILE LEfSe output file
OUTPUT_FILE the file for the output (the zip file if an archive is
required, the output directory otherwise)
optional arguments:
-h, --help show this help message and exit
--width WIDTH
--height HEIGHT
--top TOP set maximum y limit (-1.0 means automatic limit)
--bot BOT set minimum y limit (default 0.0, -1.0 means automatic
limit)
--title_font_size TITLE_FONT_SIZE
--class_font_size CLASS_FONT_SIZE
--class_label_pos {
up,down}
--subcl_mean {
y,n}
--subcl_median {
y,n}
--font_size FONT_SIZE
-n flt unused
--format {
png,pdf,svg}
the format for the output file
-f {
all,diff,one} wheter to plot all features (all), only those
differentially abundant according to LEfSe or only one
(the one given with --feature_name)
--feature_name FEATURE_NAME
The name of the feature to plot (levels separated by
.)
--feature_num FEATURE_NUM
The number of the feature to plot
--archive {
zip,none}
--background_color {
k,w}
set the color of the background
--dpi DPI
绘制系统发育树图
可以将差异结果,结合物种进化关系,绘制系统发育树图。前提是输入文件必须指定物种所有的物种等级。例如metaphlan的输出结果。如果物种太多,可以进一步对差异分析结果进行筛选过滤,只对差异特征进行绘图。
## 可直接绘制单个水平的图,也可将所有水平进行分析后,得到的结果进行绘图
## 如果很多特征都是没有组间差异的,可以对特征进行筛选后再绘图 grep -v '-' 即可
## --clade_sep 默认为1.5,但绘图时会自动调整,也可以自己设定
## --abrv_stop_lev 7 默认为5,即展示到第5水平,可设置到7,暂时到第7水平的结果。可以结合标签参数和缩写参数来展示。
lefse-plot_cladogram.py --dpi 500 all_level_lefse_significant.res all_level_lefse_significant_cladogram.pdf --format pdf --clade_sep 0.05 --abrv_stop_lev 7
详细参数:
usage: lefse-plot_cladogram.py [-h] [--clade_sep CLADE_SEP]
[--max_lev MAX_LEV]
[--max_point_size MAX_POINT_SIZE]
[--min_point_size MIN_POINT_SIZE]
[--point_edge_width MARKEREDGEWIDTH]
[--siblings_connector_width SIBLINGS_CONNECTOR_WIDTH]
[--parents_connector_width PARENTS_CONNECTOR_WIDTH]
[--radial_start_lev RADIAL_START_LEV]
[--labeled_start_lev LABELED_START_LEV]
[--labeled_stop_lev LABELED_STOP_LEV]
[--abrv_start_lev ABRV_START_LEV]
[--abrv_stop_lev ABRV_STOP_LEV]
[--expand_void_lev EXPAND_VOID_LEV]
[--class_legend_vis CLASS_LEGEND_VIS]
[--colored_connector COLORED_CONNECTORS]
[--alpha ALPHA] [--title TITLE]
[--sub_clade SUB_CLADE]
[--title_font_size TITLE_FONT_SIZE]
[--right_space_prop R_PROP]
[--left_space_prop L_PROP]
[--label_font_size LABEL_FONT_SIZE]
[--background_color {
k,w}]
[--colored_labels {
0,1}]
[--class_legend_font_size CLASS_LEGEND_FONT_SIZE]
[--dpi DPI] [--format {
png,svg,pdf}]
[--all_feats ALL_FEATS]
INPUT_FILE OUTPUT_FILE
Cladoplot
positional arguments:
INPUT_FILE tab delimited input file
OUTPUT_FILE the file for the output image
optional arguments:
-h, --help show this help message and exit
--clade_sep CLADE_SEP
--max_lev MAX_LEV
--max_point_size MAX_POINT_SIZE
--min_point_size MIN_POINT_SIZE
--point_edge_width MARKEREDGEWIDTH
--siblings_connector_width SIBLINGS_CONNECTOR_WIDTH
--parents_connector_width PARENTS_CONNECTOR_WIDTH
--radial_start_lev RADIAL_START_LEV
--labeled_start_lev LABELED_START_LEV
--labeled_stop_lev LABELED_STOP_LEV
--abrv_start_lev ABRV_START_LEV
--abrv_stop_lev ABRV_STOP_LEV
--expand_void_lev EXPAND_VOID_LEV
--class_legend_vis CLASS_LEGEND_VIS
--colored_connector COLORED_CONNECTORS
--alpha ALPHA
--title TITLE
--sub_clade SUB_CLADE
--title_font_size TITLE_FONT_SIZE
--right_space_prop R_PROP
--left_space_prop L_PROP
--label_font_size LABEL_FONT_SIZE
--background_color {
k,w}
set the color of the background
--colored_labels {
0,1}
draw the label with class color (1) or in black (0)
--class_legend_font_size CLASS_LEGEND_FONT_SIZE
--dpi DPI
--format {
png,svg,pdf}
the format for the output file
--all_feats ALL_FEATS
参考资料
https://github.com/SegataLab/lefse