- UI学习——cell的复用和自定义cell
Magnetic_h
ui学习
目录cell的复用手动(非注册)自动(注册)自定义cellcell的复用在iOS开发中,单元格复用是一种提高表格(UITableView)和集合视图(UICollectionView)滚动性能的技术。当一个UITableViewCell或UICollectionViewCell首次需要显示时,如果没有可复用的单元格,则视图会创建一个新的单元格。一旦这个单元格滚动出屏幕,它就不会被销毁。相反,它被添
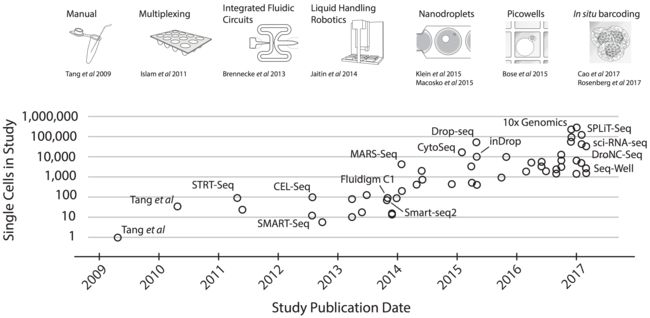
- Cell Insight | 单细胞测序技术又一新发现,可用于HIV-1和Mtb共感染个体诊断
尐尐呅
结核病是艾滋病合并其他疾病中导致患者死亡的主要原因。其中结核病由结核分枝杆菌(Mycobacteriumtuberculosis,Mtb)感染引起,获得性免疫缺陷综合症(艾滋病)由人免疫缺陷病毒(Humanimmunodeficiencyvirustype1,HIV-1)感染引起。国家感染性疾病临床医学研究中心/深圳市第三人民医院张国良团队携手深圳华大生命科学研究院吴靓团队,共同研究得出单细胞测序
- WPF中的ComboBox控件几种数据绑定的方式
互联网打工人no1
wpfc#
一、用字典给ItemsSource赋值(此绑定用的地方很多,建议熟练掌握)在XMAL中:在CS文件中privatevoidBindData(){DictionarydicItem=newDictionary();dicItem.add(1,"北京");dicItem.add(2,"上海");dicItem.add(3,"广州");cmb_list.ItemsSource=dicItem;cmb_l
- 将cmd中命令输出保存为txt文本文件
落难Coder
Windowscmdwindow
最近深度学习本地的训练中我们常常要在命令行中运行自己的代码,无可厚非,我们有必要保存我们的炼丹结果,但是复制命令行输出到txt是非常麻烦的,其实Windows下的命令行为我们提供了相应的操作。其基本的调用格式就是:运行指令>输出到的文件名称或者具体保存路径测试下,我打开cmd并且ping一下百度:pingwww.baidu.com>./data.txt看下相同目录下data.txt的输出:如果你再
- 在一台Ubuntu计算机上构建Hyperledger Fabric网络
落叶无声9
区块链超级账本Hyperledgerfabric区块链ubuntu构建hyperledgerfabric
在一台Ubuntu计算机上构建HyperledgerFabric网络Hyperledgerfabric是一个开源的区块链应用程序平台,为开发基于区块链的应用程序提供了一个起点。当我们提到HyperledgerFabric网络时,我们指的是使用HyperledgerFabric的正在运行的系统。即使只使用最少数量的组件,部署Fabric网络也不是一件容易的事。Fabric社区创建了一个名为Cello
- pyecharts——绘制柱形图折线图
2224070247
信息可视化pythonjava数据可视化
一、pyecharts概述自2013年6月百度EFE(ExcellentFrontEnd)数据可视化团队研发的ECharts1.0发布到GitHub网站以来,ECharts一直备受业界权威的关注并获得广泛好评,成为目前成熟且流行的数据可视化图表工具,被应用到诸多数据可视化的开发领域。Python作为数据分析领域最受欢迎的语言,也加入ECharts的使用行列,并研发出方便Python开发者使用的数据
- Linux MariaDB使用OpenSSL安装SSL证书
Meta39
MySQLOracleMariaDBLinuxWindowsssllinuxmariadb
进入到证书存放目录,批量删除.pem证书警告:确保已经进入到证书存放目录find.-typef-iname\*.pem-delete查看是否安装OpenSSLopensslversion没有则安装yuminstallopensslopenssl-devel开启SSL编辑/etc/my.cnf文件(没有的话就创建,但是要注意,在/etc/my.cnf.d/server.cnf配置了datadir的,
- 网络编程基础
记得开心一点啊
网络
目录♫什么是网络编程♫Socket套接字♪什么是Socket套接字♪数据报套接字♪流套接字♫数据报套接字通信模型♪数据报套接字通讯模型♪DatagramSocket♪DatagramPacket♪实现UDP的服务端代码♪实现UDP的客户端代码♫流套接字通信模型♪流套接字通讯模型♪ServerSocket♪Socket♪实现TCP的服务端代码♪实现TCP的客户端代码♫什么是网络编程网络编程,指网络上
- K近邻算法_分类鸢尾花数据集
_feivirus_
算法机器学习和数学分类机器学习K近邻
importnumpyasnpimportpandasaspdfromsklearn.datasetsimportload_irisfromsklearn.model_selectionimporttrain_test_splitfromsklearn.metricsimportaccuracy_score1.数据预处理iris=load_iris()df=pd.DataFrame(data=ir
- 4.C_数据结构_队列
荣世蓥
数据结构数据结构
概述什么是队列:队列是限定在两端进行插入操作和删除操作的线性表。具有先入先出(FIFO)的特点相关名词:队尾:写入数据的一段队头:读取数据的一段空队:队列中没有数据,队头指针=队尾指针满队:队列中存满了数据,队尾指针+1=队头指针循环队列1、基本内容循环队列是以数组形式构成的队列数据结构。循环队列的结构体如下:typedefintdata_t;//队列数据类型#defineN64//队列容量typ
- vue项目element-ui的table表格单元格合并
酋长哈哈
vue.jselementuijavascript前端
一、合并效果二全部代码exportdefault{name:'CellMerge',data(){return{tableData:[{id:'1',name:'王小虎',amount1:'165',amount2:'3.2',amount3:10},{id:'1',name:'王小虎',amount1:'162',amount2:'4.43',amount3:12},{id:'1',name:'
- python tif转png
Python与遥感
python开发语言
importosfromosgeoimportgdalimportnumpyasnpfromPILimportImage#提取432三波段fromspectralimport*#输入文件夹路径defget_img(dataset_img):width=dataset_img.RasterXSize#获取行列数height=dataset_img.RasterYSizebands=dataset_i
- MongoDB知识概括
GeorgeLin98
持久层mongodb
MongoDB知识概括MongoDB相关概念单机部署基本常用命令索引-IndexSpirngDataMongoDB集成副本集分片集群安全认证MongoDB相关概念业务应用场景:传统的关系型数据库(如MySQL),在数据操作的“三高”需求以及应对Web2.0的网站需求面前,显得力不从心。解释:“三高”需求:①Highperformance-对数据库高并发读写的需求。②HugeStorage-对海量数
- Vue中table合并单元格用法
weixin_30613343
javascriptViewUI
地名结果人名性别{{item.name}}已完成未完成{{item.groups[0].name}}{{item.groups[0].sex}}{{item.groups[son].name}}{{item.groups[son].sex}}exportdefault{data(){return{list:[{name:'地名1',result:'1',groups:[{name:'张三',sex
- uniapp map组件自定义markers标记点
以对_
uni-app学习记录uni-appjavascript前端
需求是根据后端返回数据在地图上显示标记点,并且根据数据状态控制标记点颜色,标记点背景通过两张图片实现控制{{item.options.labelName}}exportdefault{data(){return{storeIndex:0,locaInfo:{longitude:120.445172,latitude:36.111387},markers:[//标点列表{id:1,//标记点idin
- 博客网站制作教程
2401_85194651
javamaven
首先就是技术框架:后端:Java+SpringBoot数据库:MySQL前端:Vue.js数据库连接:JPA(JavaPersistenceAPI)1.项目结构blog-app/├──backend/│├──src/main/java/com/example/blogapp/││├──BlogApplication.java││├──config/│││└──DatabaseConfig.java
- vue + Element UI table动态合并单元格
我家媳妇儿萌哒哒
elementUIvue.js前端javascript
一、功能需求1、根据名称相同的合并工作阶段和主要任务合并这两列,但主要任务内容一样,但要考虑主要任务一样,但工作阶段不一样的情况。(枞向合并)2、落实情况里的定量内容和定性内容值一样则合并。(横向合并)二、功能实现exportdefault{data(){return{tableData:[{name:'a',address:'1',age:'1',six:'2'},{name:'a',addre
- Python实现TIFF 文件转换为 PNG 和 JPG 格式
sand&wich
python开发语言
在日常的图像处理工作中,可能会遇到需要将TIFF格式的图像转换为其他格式的情况,例如PNG和JPG。下面,本文将介绍如何使用Python和GDAL库实现这一功能。准备工作在开始之前,请确保已经安装了必要的库:GDAL(GeospatialDataAbstractionLibrary)可以使用以下命令安装GDAL:pipinstallgdal代码实现以下是一个将TIFF文件转换为PNG文件的示例代码
- 使用datepicker和uploadify的冲突解决(IE双击才能打开附件上传对话框)
zhanglb12
在开发的过程当中,IE的兼容无疑是我们的一块绊脚石,在我们使用的如期的datepicker插件和使用上传附件的uploadify插件的时候,两者就产生冲突,只要点击过时间的插件,uploadify上传框要双才能打开ie浏览器提示错误Missinginstancedataforthisdatepicker解决方案//if(.browser.msie&&'9.0'===.browser.version
- golang获取用户输入的几种方式
余生逆风飞翔
golang开发语言后端
一、定义结构体typeUserInfostruct{Namestring`json:"name"`Ageint`json:"age"`Addstring`json:"add"`}typeReturnDatastruct{Messagestring`json:"message"`Statusstring`json:"status"`DataUserInfo`json:"data"`}二、get请求的
- leetcode-124 Binary Tree Maximum Path Sum
乐观的大鹏
LeetCode
Givenanon-emptybinarytree,findthemaximumpathsum.Forthisproblem,apathisdefinedasanysequenceofnodesfromsomestartingnodetoanynodeinthetreealongtheparent-childconnections.Thepathmustcontainatleastonenodea
- LeetCode 673. Number of Longest Increasing Subsequence (Java版; Meidum)
littlehaes
字符串动态规划算法leetcode数据结构
welcometomyblogLeetCode673.NumberofLongestIncreasingSubsequence(Java版;Meidum)题目描述Givenanunsortedarrayofintegers,findthenumberoflongestincreasingsubsequence.Example1:Input:[1,3,5,4,7]Output:2Explanatio
- 【Java】已解决:org.springframework.jdbc.datasource.lookup.DataSourceLookupFailureException
屿小夏
java开发语言
文章目录一、分析问题背景问题背景描述出现问题的场景二、可能出错的原因三、错误代码示例四、正确代码示例五、注意事项已解决:org.springframework.jdbc.datasource.lookup.DataSourceLookupFailureException在使用Spring框架进行开发时,数据源的配置和使用是非常关键的一环。然而,有时候我们可能会遇到org.springframewo
- el-table实现全选整表,单元一页复选框功能
周bro
vue.jselementuijavascript前端
全选整表单选一页0":popper-append-to-body="false":total="tableData.length":page-size="pageObj.pagesize":page-sizes="[10,50,100]"layout="total,sizes,prev,pager,next"@size-change="handleSizeChange"@current-chang
- Vue + Express实现一个表单提交
九旬大爷的梦
最近在折腾一个cms系统,用的vue+express,但是就一个表单提交就弄了好久,记录一下。环境:Node10+前端:Vue服务端:Express依赖包:vueexpressaxiosexpress-formidableelement-ui(可选)前言:axiosget请求参数是:paramsaxiospost请求参数是:dataexpressget接受参数是req.queryexpresspo
- iOS下拉放大效果
RobinZhao
好多下拉放大的实现方式是在tableView上面添加一个view,同时更改tableView.contentInset,下拉时改变view的frame来实现。今天用另外一种方式实现,效果如下:加油.gif具体实现方法:通过tableViewCell结合xib来实现,具体代码如下HomeViewController.m代码#import"HomeViewController.h"#import"He
- vant-element-ts一起使用存在的问题
flynn_
问题总结vue
由于vant-ui与element-ui部分组件存在冲突,导致在vue-typescript中出现错误:Subsequentpropertydeclarationsmusthavethesametype.Property'$notify'mustbeoftype'ElNotification',butherehastype'Notify'.方案:一个全局导入,一个按需导入,避免冲突的组件同时使用,
- Kubernetes部署MySQL数据持久化
沫殇-MS
KubernetesMySQL数据库kubernetesmysql容器
一、安装配置NFS服务端1、安装nfs-kernel-server:sudoapt-yinstallnfs-kernel-server2、服务端创建共享目录#列出所有可用块设备的信息lsblk#格式化磁盘sudomkfs-text4/dev/sdb#创建一个目录:sudomkdir-p/data/nfs/mysql#更改目录权限:sudochown-Rnobody:nogroup/data/nfs
- Hadoop架构
henan程序媛
hadoop大数据分布式
一、案列分析1.1案例概述现在已经进入了大数据(BigData)时代,数以万计用户的互联网服务时时刻刻都在产生大量的交互,要处理的数据量实在是太大了,以传统的数据库技术等其他手段根本无法应对数据处理的实时性、有效性的需求。HDFS顺应时代出现,在解决大数据存储和计算方面有很多的优势。1.2案列前置知识点1.什么是大数据大数据是指无法在一定时间范围内用常规软件工具进行捕捉、管理和处理的大量数据集合,
- Scanpy源码浅析之pp.normalize_total
何物昂
版本导入Scanpy,其版本为'1.9.1',如果你看到的源码和下文有差异,其可能是由于版本差异。importscanpyasscsc.__version__#'1.9.1'例子函数pp.normalize_total用于Normalizecountspercell,其源代码在scanpy/preprocessing/_normalization.py我们通过一个简单例子来了解该函数主要功能:将一
- mongodb3.03开启认证
21jhf
mongodb
下载了最新mongodb3.03版本,当使用--auth 参数命令行开启mongodb用户认证时遇到很多问题,现总结如下:
(百度上搜到的基本都是老版本的,看到db.addUser的就是,请忽略)
Windows下我做了一个bat文件,用来启动mongodb,命令行如下:
mongod --dbpath db\data --port 27017 --directoryperdb --logp
- 【Spark103】Task not serializable
bit1129
Serializable
Task not serializable是Spark开发过程最令人头疼的问题之一,这里记录下出现这个问题的两个实例,一个是自己遇到的,另一个是stackoverflow上看到。等有时间了再仔细探究出现Task not serialiazable的各种原因以及出现问题后如何快速定位问题的所在,至少目前阶段碰到此类问题,没有什么章法
1.
package spark.exampl
- 你所熟知的 LRU(最近最少使用)
dalan_123
java
关于LRU这个名词在很多地方或听说,或使用,接下来看下lru缓存回收的实现
1、大体的想法
a、查询出最近最晚使用的项
b、给最近的使用的项做标记
通过使用链表就可以完成这两个操作,关于最近最少使用的项只需要返回链表的尾部;标记最近使用的项,只需要将该项移除并放置到头部,那么难点就出现 你如何能够快速在链表定位对应的该项?
这时候多
- Javascript 跨域
周凡杨
JavaScriptjsonp跨域cross-domain
- linux下安装apache服务器
g21121
apache
安装apache
下载windows版本apache,下载地址:http://httpd.apache.org/download.cgi
1.windows下安装apache
Windows下安装apache比较简单,注意选择路径和端口即可,这里就不再赘述了。 2.linux下安装apache:
下载之后上传到linux的相关目录,这里指定为/home/apach
- FineReport的JS编辑框和URL地址栏语法简介
老A不折腾
finereportweb报表报表软件语法总结
JS编辑框:
1.FineReport的js。
作为一款BS产品,browser端的JavaScript是必不可少的。
FineReport中的js是已经调用了finereport.js的。
大家知道,预览报表时,报表servlet会将cpt模板转为html,在这个html的head头部中会引入FineReport的js,这个finereport.js中包含了许多内置的fun
- 根据STATUS信息对MySQL进行优化
墙头上一根草
status
mysql 查看当前正在执行的操作,即正在执行的sql语句的方法为:
show processlist 命令
mysql> show global status;可以列出MySQL服务器运行各种状态值,我个人较喜欢的用法是show status like '查询值%';一、慢查询mysql> show variab
- 我的spring学习笔记7-Spring的Bean配置文件给Bean定义别名
aijuans
Spring 3
本文介绍如何给Spring的Bean配置文件的Bean定义别名?
原始的
<bean id="business" class="onlyfun.caterpillar.device.Business">
<property name="writer">
<ref b
- 高性能mysql 之 性能剖析
annan211
性能mysqlmysql 性能剖析剖析
1 定义性能优化
mysql服务器性能,此处定义为 响应时间。
在解释性能优化之前,先来消除一个误解,很多人认为,性能优化就是降低cpu的利用率或者减少对资源的使用。
这是一个陷阱。
资源时用来消耗并用来工作的,所以有时候消耗更多的资源能够加快查询速度,保持cpu忙绿,这是必要的。很多时候发现
编译进了新版本的InnoDB之后,cpu利用率上升的很厉害,这并不
- 主外键和索引唯一性约束
百合不是茶
索引唯一性约束主外键约束联机删除
目标;第一步;创建两张表 用户表和文章表
第二步;发表文章
1,建表;
---用户表 BlogUsers
--userID唯一的
--userName
--pwd
--sex
create
- 线程的调度
bijian1013
java多线程thread线程的调度java多线程
1. Java提供一个线程调度程序来监控程序中启动后进入可运行状态的所有线程。线程调度程序按照线程的优先级决定应调度哪些线程来执行。
2. 多数线程的调度是抢占式的(即我想中断程序运行就中断,不需要和将被中断的程序协商)
a)
- 查看日志常用命令
bijian1013
linux命令unix
一.日志查找方法,可以用通配符查某台主机上的所有服务器grep "关键字" /wls/applogs/custom-*/error.log
二.查看日志常用命令1.grep '关键字' error.log:在error.log中搜索'关键字'2.grep -C10 '关键字' error.log:显示关键字前后10行记录3.grep '关键字' error.l
- 【持久化框架MyBatis3一】MyBatis版HelloWorld
bit1129
helloworld
MyBatis这个系列的文章,主要参考《Java Persistence with MyBatis 3》。
样例数据
本文以MySQL数据库为例,建立一个STUDENTS表,插入两条数据,然后进行单表的增删改查
CREATE TABLE STUDENTS
(
stud_id int(11) NOT NULL AUTO_INCREMENT,
- 【Hadoop十五】Hadoop Counter
bit1129
hadoop
1. 只有Map任务的Map Reduce Job
File System Counters
FILE: Number of bytes read=3629530
FILE: Number of bytes written=98312
FILE: Number of read operations=0
FILE: Number of lar
- 解决Tomcat数据连接池无法释放
ronin47
tomcat 连接池 优化
近段时间,公司的检测中心报表系统(SMC)的开发人员时不时找到我,说用户老是出现无法登录的情况。前些日子因为手头上 有Jboss集群的测试工作,发现用户不能登录时,都是在Tomcat中将这个项目Reload一下就好了,不过只是治标而已,因为大概几个小时之后又会 再次出现无法登录的情况。
今天上午,开发人员小毛又找到我,要我协助将这个问题根治一下,拖太久用户难保不投诉。
简单分析了一
- java-75-二叉树两结点的最低共同父结点
bylijinnan
java
import java.util.LinkedList;
import java.util.List;
import ljn.help.*;
public class BTreeLowestParentOfTwoNodes {
public static void main(String[] args) {
/*
* node data is stored in
- 行业垂直搜索引擎网页抓取项目
carlwu
LuceneNutchHeritrixSolr
公司有一个搜索引擎项目,希望各路高人有空来帮忙指导,谢谢!
这是详细需求:
(1) 通过提供的网站地址(大概100-200个网站),网页抓取程序能不断抓取网页和其它类型的文件(如Excel、PDF、Word、ppt及zip类型),并且程序能够根据事先提供的规则,过滤掉不相干的下载内容。
(2) 程序能够搜索这些抓取的内容,并能对这些抓取文件按照油田名进行分类,然后放到服务器不同的目录中。
- [通讯与服务]在总带宽资源没有大幅增加之前,不适宜大幅度降低资费
comsci
资源
降低通讯服务资费,就意味着有更多的用户进入,就意味着通讯服务提供商要接待和服务更多的用户,在总体运维成本没有由于技术升级而大幅下降的情况下,这种降低资费的行为将导致每个用户的平均带宽不断下降,而享受到的服务质量也在下降,这对用户和服务商都是不利的。。。。。。。。
&nbs
- Java时区转换及时间格式
Cwind
java
本文介绍Java API 中 Date, Calendar, TimeZone和DateFormat的使用,以及不同时区时间相互转化的方法和原理。
问题描述:
向处于不同时区的服务器发请求时需要考虑时区转换的问题。譬如,服务器位于东八区(北京时间,GMT+8:00),而身处东四区的用户想要查询当天的销售记录。则需把东四区的“今天”这个时间范围转换为服务器所在时区的时间范围。
- readonly,只读,不可用
dashuaifu
jsjspdisablereadOnlyreadOnly
readOnly 和 readonly 不同,在做js开发时一定要注意函数大小写和jsp黄线的警告!!!我就经历过这么一件事:
使用readOnly在某些浏览器或同一浏览器不同版本有的可以实现“只读”功能,有的就不行,而且函数readOnly有黄线警告!!!就这样被折磨了不短时间!!!(期间使用过disable函数,但是发现disable函数之后后台接收不到前台的的数据!!!)
- LABjs、RequireJS、SeaJS 介绍
dcj3sjt126com
jsWeb
LABjs 的核心是 LAB(Loading and Blocking):Loading 指异步并行加载,Blocking 是指同步等待执行。LABjs 通过优雅的语法(script 和 wait)实现了这两大特性,核心价值是性能优化。LABjs 是一个文件加载器。RequireJS 和 SeaJS 则是模块加载器,倡导的是一种模块化开发理念,核心价值是让 JavaScript 的模块化开发变得更
- [应用结构]入口脚本
dcj3sjt126com
PHPyii2
入口脚本
入口脚本是应用启动流程中的第一环,一个应用(不管是网页应用还是控制台应用)只有一个入口脚本。终端用户的请求通过入口脚本实例化应用并将将请求转发到应用。
Web 应用的入口脚本必须放在终端用户能够访问的目录下,通常命名为 index.php,也可以使用 Web 服务器能定位到的其他名称。
控制台应用的入口脚本一般在应用根目录下命名为 yii(后缀为.php),该文
- haoop shell命令
eksliang
hadoophadoop shell
cat
chgrp
chmod
chown
copyFromLocal
copyToLocal
cp
du
dus
expunge
get
getmerge
ls
lsr
mkdir
movefromLocal
mv
put
rm
rmr
setrep
stat
tail
test
text
- MultiStateView不同的状态下显示不同的界面
gundumw100
android
只要将指定的view放在该控件里面,可以该view在不同的状态下显示不同的界面,这对ListView很有用,比如加载界面,空白界面,错误界面。而且这些见面由你指定布局,非常灵活。
PS:ListView虽然可以设置一个EmptyView,但使用起来不方便,不灵活,有点累赘。
<com.kennyc.view.MultiStateView xmlns:android=&qu
- jQuery实现页面内锚点平滑跳转
ini
JavaScripthtmljqueryhtml5css
平时我们做导航滚动到内容都是通过锚点来做,刷的一下就直接跳到内容了,没有一丝的滚动效果,而且 url 链接最后会有“小尾巴”,就像#keleyi,今天我就介绍一款 jquery 做的滚动的特效,既可以设置滚动速度,又可以在 url 链接上没有“小尾巴”。
效果体验:http://keleyi.com/keleyi/phtml/jqtexiao/37.htmHTML文件代码:
&
- kafka offset迁移
kane_xie
kafka
在早前的kafka版本中(0.8.0),offset是被存储在zookeeper中的。
到当前版本(0.8.2)为止,kafka同时支持offset存储在zookeeper和offset manager(broker)中。
从官方的说明来看,未来offset的zookeeper存储将会被弃用。因此现有的基于kafka的项目如果今后计划保持更新的话,可以考虑在合适
- android > 搭建 cordova 环境
mft8899
android
1 , 安装 node.js
http://nodejs.org
node -v 查看版本
2, 安装 npm
可以先从 https://github.com/isaacs/npm/tags 下载 源码 解压到
- java封装的比较器,比较是否全相同,获取不同字段名字
qifeifei
非常实用的java比较器,贴上代码:
import java.util.HashSet;
import java.util.List;
import java.util.Set;
import net.sf.json.JSONArray;
import net.sf.json.JSONObject;
import net.sf.json.JsonConfig;
i
- 记录一些函数用法
.Aky.
位运算PHP数据库函数IP
高手们照旧忽略。
想弄个全天朝IP段数据库,找了个今天最新更新的国内所有运营商IP段,copy到文件,用文件函数,字符串函数把玩下。分割出startIp和endIp这样格式写入.txt文件,直接用phpmyadmin导入.csv文件的形式导入。(生命在于折腾,也许你们觉得我傻X,直接下载人家弄好的导入不就可以,做自己的菜鸟,让别人去说吧)
当然用到了ip2long()函数把字符串转为整型数
- sublime text 3 rust
wudixiaotie
Sublime Text
1.sublime text 3 => install package => Rust
2.cd ~/.config/sublime-text-3/Packages
3.mkdir rust
4.git clone https://github.com/sp0/rust-style
5.cd rust-style
6.cargo build --release
7.ctrl