三月week1文献阅读:
Pathway enrichment analysis and visualzation of omics data using g:Profiler,GSEA,Cytoscape and EnrichmentMap
通路富集分析和可视化组学的软件数据使用:Profiler,GSEA,Cytoscape and EnrichmentMap
摘要
通路富集分析帮助研究人员在从基因组规模(组学)实验中产生的基因列表获得力学洞察力。这种方法超出了随机情况下的预期,它可以识别出富集在基因列表中生物通路。作者们解释了通路富集分析的步骤,并提出了一个实用的逐步指南,以帮助解释RNA-seq和基因组测序实验产生的基因列表。该方案包括三个主要步骤:
- 从组学数据定义基因列表,
- 富集通路的统计确定,
- 可视化和解释结果。
描述了如何使用这个方案与已发表的差异表达基因和突变的癌症基因例子结合;进而,这些方案可以应用于不同类型的组学数据。该方案描述了可视化技术的创新,提供全面的背景和故障排除指南,并使用免费和经常更新的软件,包括g:Profiler, Gene Set Enrichment Analysis (GSEA), Cytoscape和EnrichmentMap。完整的方案可以在大约4.5小时内完成,并且是为没有经过生物信息学培训的生物学家设计的。
介绍 Indroduction
生物样品中DNA、RNA和蛋白质的全面的综合的定量现已成为常规。结果数据呈指数增长,他们的分析帮助研究人员发现新的生物功能、基因型-表型关系和疾病机制。然而,对这些数据的分析和解释对许多研究人员来说是一个重大挑战。分析常常导致一长串的基因,需要不切实际的大量手工文献搜索来解释。解决这个问题的标准方法是通路富集分析,它将大的基因列表总结为更容易解释的通路的小列表。使用几种常见的统计检验方法,考虑实验中检测到的基因数量、它们的相对排名以及注释到感兴趣的通路的基因数量,对实验基因列表中相对于预期的过度表达进行统计检验。例如,含有40%细胞周期基因的实验数据惊人地丰富,因为只有8%的人类蛋白编码基因参与了这一过程。
在最近的一个例子中,我们使用通路富集分析来帮助确定polycomb repressive complex (PRC2)的组蛋白和DNA甲基化作为室管膜瘤(最常见的儿童期脑癌之一)的第一个合理治疗靶点。现有的药物如5-氮杂替丁(5-azacytidine)可以靶向这一通路。5-氮杂替丁曾在一位晚期病人身上出于同情心使用,并阻止了快速转移性肿瘤的生长。在另一个例子中,我们分析了自闭症中罕见的拷贝数变异(CNVs),并确定了几个受基因缺失影响的重要通路,而通过对单个基因或loci的病例对照关联测试,几乎没有发现显著的命中。这些例子说明了利用通路富集分析可以获得的对生物机制的有用见解。
方案发展Development of the protocol
该方案是为那些对解释组学数据感兴趣的实验生物学家设计的。它只需要学习和使用“点击”计算机软件的能力,但是高级用户可以从我们作为补充方案1-4提供的自动分析脚本中受益。我们以之前发表的人类基因表达和体细胞突变数据为例进行分析;然而,我们的概念框架适用于分析来自大规模数据的任何生物体的基因或生物分子列表,包括蛋白质组学、基因组学、表观基因组学和基因调控研究。我们在许多项目中广泛使用通路富集分析,并评估了许多可用的工具。我们在这里介绍的软件包是根据它们的易用性、免费访问、高级功能、广泛的文档和最新的数据库而选择的,它们是我们在研究中每天使用的软件包,并向合作者和学生推荐。此外,我们还向这些工具的开发人员提供了反馈,允许他们实现我们在发布的分析中需要的特性。这些工具是g:Profiler, GSEA, Cytoscape和EnrichmentMap,都可以在网上免费获得:
●g:分析器(https://biit.cs.ut.ee/gprofiler/)
●GSEA(http://software.broadinstitute.org/gsea/)
●Cytoscape (http://www.cytoscape.org/)
●EnrichmentMap(http://www.baderlab.org/Software/EnrichmentMap)
流程概述 Overview of the procedure
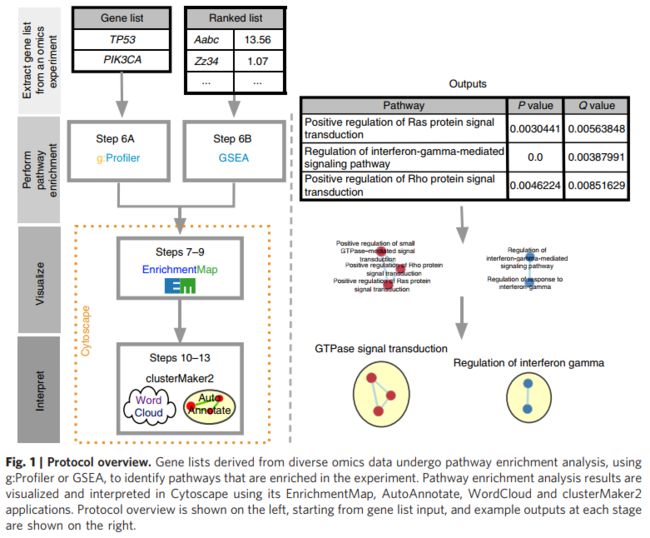
本节概述了通路富集分析的主要阶段。下面的过程提供了详细的分步协议。通路富集分析包括三个主要阶段(fig1;基本定义见Box1)
- 使用组学数据定义感兴趣的基因列表。组学实验在实验环境中全面测量基因的活性。考虑到实验设计,得到的原始数据集通常需要计算处理,如标准化和评分,以识别感兴趣的基因。例如,可以从RNA-seq数据得到两组样本间差异表达的基因列表。其他类型的组学实验,如基因表达微阵列、定量蛋白组学、种系和体细胞基因组测序、全球DNA甲基化分析等,均可用于本方案;然而,每种类型的数据可能需要特定的预处理步骤(参见“与其他方法的比较”一节)。
| Box 1 | Definitons |
|---|
| Pathway: 共同起作用以进行生物过程的基因 |
| Gene Set:包括通路中的所有基因。基因集可以基于基因之间的各种关系,如细胞定位(如核基因)或酶功能(如蛋白激酶)。不包括蛋白质相互作用等细节。 |
| Gene list of interest:感兴趣的基因列表。从组学实验中获得的基因列表,用于通路富集分析 |
| Ranked gene list:在许多组学数据(如RNA-seq基因表达数据)中,可以根据一定的评分(如差异表达水平)对基因进行排序,为通路富集分析提供更多信息。与在排序列表中随机分布的路径基因相比,在排序列表顶部聚集的基因富集的路径得分更高 |
| Pathway enrichment analysis:一种统计技术,用于识别在基因列表或感兴趣的排序基因列表中显著表示的通路 |
| Multiple testing correction:成千上万的通路可以单独进行富集试验,这可能导致显著富集P值,单独出现。多次测试校正是一种统计技术纠正个别富集试验的P值,以解决这个问题,并减少假阳性富集的机会(Box 3)。 |
| Leading-edge gene:在GSEA分析中,在最大值或最大值之前的排序中发现的基因子集。这部分基因通常被定义为富集通路。 |
2.通路富集分析:统计方法用于识别从第1阶段开始,相对于随机预期的基因列表中富集的通路。对给定数据库中的所有通路进行富集基因列表测试(参见Box2中的通路数据库列表)。几种已建立的通路富集分析方法是可用的,使用哪一种方法取决于基因列表的类型(参见“与其他方法的比较”一节)。
3.通路富集分析结果的可视化和解释。许多富集的通路可以在阶段中被识别,通常包括同一通路的相关版本。可视化可以帮助识别主要的生物学主题及其相互关系,以便进行深入的研究和实验评价。
| Box 2 | Pathway enrichment analysis resources |
|---|
| Pathway data bases:(通路数据库)我们列出了一些大型的、开放的和方便访问的通路数据库,它们为通路富集分析提供了最大的价值。有数百个pathway数据库可供多种用途。 |
| Gene set databases:(Gene set databases) |
| ●Gene Ontology(GO):GO提供了一套分层次组织成千上万的生物过程,标准化的术语分子功能和细胞组件,以及根据这些术语为多个物种策划和预测基因注释。生物过程GO注释是通路富集分析中最常用的资源。 ●分子标记数据库(MSigDB): MSigDB是一个基于GO、通路、管理、个体组学研究、序列基序、染色体位置、致癌和免疫表达标记以及GSEA团队维护的各种计算分析的基因集数据库(http://www.msigdb.org)。一个相对不冗余的“hallmark”基因集集合是可用的。该数据可用于多种通路富集方法 |
| Detailed biochemical pathway databases:(详细生化通路数据库)这些数据库由一组管理员维护,他们手工收集详细的通路信息,包括生化反应、基因调控事件和其他基因相互作用。信息可以导出或转换为基因集格式。 |
| ●Reactome:人类最积极更新通用公共数据库路径(http://www.reactome.org)。 |
| ●Panther:人类信号通路(http://pantherdb.org/pathway)。 |
| ●NetPath:人类与关注癌症信号通路和免疫学(http://www.netpath.org/)。 |
| ●HumanCyc:人类代谢通路(http://humancyc.org/)。 |
| Pathway meta-databases:这些数据库从多个原始路径数据库中收集详细的路径描述。 |
| ●Pathway Commons:从其他通路收集信息数据库,并提供标准化的格式(http://www.pathwaycommons.org)。 |
| ●WikiPathways:社区驱动的通路,还包括与其他数据库通路(http://www.wikipathways.org/) |
| Pathway meta-databases:(元数据库通路)这些数据库从多个原始路径数据库中收集详细的路径描述。 |
| ●通路Commons45:从其他途径收集信息数据库,并提供标准化通路(http://www.pathwaycommons.org)。 |
| ●WikiPathways48:社区驱动的通路,还包括与其他数据库路径(http://www.wikipathways.org/) |
| Box 3 | Mutiple testing correction |
|---|
| 在典型的通路富集分析中,重复的统计检验仅凭偶然的机会就会得到一些显著的P值。为了纠正这一点,Mutiple testing correction (多测试校正方法)系统地降低了从一系列测试中得出的每个P值的显著性。在本方案中,g:Profiler和GSEA自动对P值进行多次测试校正。最常用的方法是hh -FDR(或简称FDR)34。它是基于一种降序程序,使用未校正的p值阈值和测试次数来估计错误富集的通路比总的富集的通路的比例。例如,假设100条通路P值<0.05富集,且FDR在P值<0.05时为5%,则认为其中5条通路可能富集错误。另外,经典的Bonferroni多重测试校正通过将显著性阈值除以测试次数来调整显著性阈值。在实际应用中,该方法将每个未校正的P值乘以所进行的试验次数,并应用显著性截止(例如,如果测试了100条通路,P值0.001将成为不显著的Q值0.1)。该技术确保选择至少一个错误富集通路的概率低于校正后的P值阈值。Bonferroni校正通常被认为对差异基因表达和通路富集分析过于保守,因为部分假阳性结果是可以容忍的。重要的是,Bonferroni和BHFDR都假设测试是独立的,而由于基因重叠和串扰,通路通常是不独立的。因此,虽然hh - fdr对通路分析的估计是不准确的,但在实际应用中,它们仍然可以用于筛选和假设生成,因此经常使用。 |
Stage1:使用组学数据定义感兴趣的基因列表 Stage1:defintion of a gene list of interest using omics data
公司实验生成的原始数据,必须获得能够处理信息适合通路富集分析(补充1和2)。具体处理步骤是特定的组学实验类型和可能的标准,因此通常直接面向实现,否则,在这种情况下,先进的计算技能可能被需要用来进行数据处理。已经建立的组学技术可以使用标准的处理方法,并且由生成数据的核心设备最方便地执行。从组学数据中定义基因列表有两种主要方法:列表或排序列表。某些组学数据自然会产生一个基因列表,例如通过外显子组测序确定的肿瘤中所有体细胞突变的基因,或者蛋白质组学实验中所有与诱饵相互作用的蛋白质。这样的列表适合使用g:Profiler直接输入通路富集分析(步骤6A)。其他组学数据自然会产生排序列表。例如,一个基因列表可以根据不同的基因进行排序在全基因组CRISPR屏幕上的表达评分或敏感性。一些通路富集分析方法是通过一个特定阈值(如fdr调整的P值<0.05和折叠改变的>2)来筛选一个排序的基因列表。其他方法,如GSEA,旨在分析所有可用基因的排序列表,不需要阈值。全基因组排序表适用于使用GSEA进行通路富集分析的输入(步骤6B)。应该使用g:Profiler分析部分(非全基因组)排序的基因列表。
作为一个例子,我们描述了对卵巢癌样本的原始RNA-seq数据的分析,以定义一个排序的基因列表。DNA序列读取是经过质量过滤的(例如,通过修剪去除低质量的碱基),并映射到基因组范围内的一组参考转录本,以支持对每个转录本的读取计数。读取计数在基因级别上进行聚合(每个基因的计数)。通常,RNA-seq数据可用于多个生物复制(三个或更多)的多个实验条件(两个或更多,例如,治疗与控制)。对所有样本的每个基因读计数进行标准化,以消除样本之间不需要的技术差异,例如,由于测序车道或每次测序运行的总读数的差异。
接下来,检测每个基因的读计数,以确定样本组之间的差异表达(例如,治疗组与对照组)(补充协议1和2分别用于RNA-seq和微阵列数据)。edgeR、DESeq、limma/voom和Cufflinks等软件包实现了RNA-seq数据规范化和差异表达分析的过程。差异基因表达分析结果包括:
(1)差异表达显著性的P值;
(2)通过使用Benjamini Hochberg假发现率(hh - fdr)程序(Box 3)对所有基因进行多次检测校正后的相关Q值(即调整后的P值);
(3)效应大小和表达方向的改变,使上调的基因为阳性,位于列表的顶部,下调的基因为阴性,位于列表的底部,常表达为log-transform fold-change。
然后基因列表根据一个或多个这些值排名(例如,−log10乘以P值的符号对数转换叠化)并使用通路富集分析研究
Stage2A阶段:利用g:Profiler对基因列表进行通路富集分析(步骤6A)Stage 2A:pathway enrichment analysis of a gene list using g:Profiler(Step 6A)
默认分析实现g:Profiler和类似的基于web的工具搜索途径的基因明显富集(即占大多数)感兴趣的基因的固定列表,与基因组中所有基因(步骤6)(Box 4)。富集的P值的通路计算使用Fisher正确概率法和多个测试校正应用(Box3)。
| Box 4 | Statistical tests in pathway enrichment analysis |
|---|
| 用于基因通路富集分析的常用统计检验是基于超几何分布的Fisher精确检验。它决定了通路中相关基因的比例是否高于通路外基因的比例(即、即背景基因集)。自该测试首次引入以来,许多改进的测试都利用了连续的实验分数,避免了应用任意阈值。我们将统计富集检测的类型分类如下: |
| 1.排序与非排序检测。排序与非排序检测。排序测试将排序基因列表作为输入,而非排序检测(如Fisher 's precision test)将感兴趣的基因列表作为输入。排序测试对于产生有意义的排序(如差异基因表达)的实验更可取,因为可以避免任意阈值。对于自然生成感兴趣的基因列表的实验(例如,癌症中的体细胞突变,与诱饵蛋白相互作用的蛋白质),非排名测试是更可取的。排名测试的例子包括在g:Profiler ' ordered查询'选项中实现的修改过的Fisher精确测试,以及在GSEA中实现的修改过的Kolmogorov-Smirnov测试。 |
| 2.精确与基于排列的测试。精确测试使用数学模型(如分布)直接计算精确的P值。基于排列的测试利用数据重采样来估计经验P值,通常表示为结果与实际数据相同或更好的排列数量,除以排列数量。例如,在病例对照研究中,我们可以将病例随机化,并将对照标记1000次,每次重复通路富集分析,以了解我们观察到相同或更强的通路富集信号的频率。排列测试可以定制以考虑特定的数据属性和偏差。如果适用的话,最好进行准确的测试,因为这些测试可以快速计算出准确的P值。然而,为特定的应用程序设计正确的精确测试可能具有挑战性,在这种情况下,自定义排列测试通常是首选的选项。 |
| 3.竞争性和独立的测试。竞争测试确定感兴趣的基因列表是否丰富通路相对于背景中所有基因集。因此,每个通路基因列表中的“竞争”富集的基因背景。相比之下,独立的测试计算统计独特通路水平,忽视基因的背景。例如,一个独立的测试可以评估在一个给定的通路内的基因表达是否在病例样本中不同于对照样本。竞争通路富集分析是目前最流行的方法,通常适用于基因表达数据。然而,如果单个基因差异不显著,需要在通路基因集水平汇集以识别信号,例如在分析罕见基因时,则必须使用自包含测试突变或其他低单基因计数的数据。在特定环境下,混合方法可能比独立测试更可取。例如,对于罕见的CNV数据,纠正对全球CNV负担的自包含检测可导致更具体的生物学结果。最后,竞争性富集测试(如Fisher’s precision test)忽略了基因之间的相关性,而修改后的竞争性测试(如Camera71)考虑到了这些因素,因此通常会产生更严格的结果(例如,参见补充方案3)。独立测试不存在这个问题。 |
| 总之,如果数据中的基因可以进行排序,那么应该使用排序测试。Fisher’s precision检验通常用于未排序的基因列表,该检验的修正版本可用于排序列表。在大多数情况下,竞争性测试是足够的,除非基因水平的信号很弱。 |
g:Profiler还包括一个有序富集测试,它适用于按分数排序的多达几千个基因的列表,而基因组中的其他基因缺乏有意义的排序信号。例如,显著突变的基因可以根据癌症驱动预测方法的得分进行排序。该分析在输入基因的更大的子列表上重复了修正的Fisher精确测试,并报告了每个通路P值最大的子列表。g:Profiler搜索一组表示基因本体论(GO)术语、通路、网络、调控元件和疾病表型的基因集。可以选择主要的基因集类别自定义搜索。
使用Fisher精确试验或相关试验的途径富集方法需要对背景基因进行定义以便进行比较。所有带注释的蛋白质编码基因通常被用作默认值。如果实验只能直接测量所有基因的一个子集,这将导致P值的不适当膨胀和假阳性结果。例如,在分析来自目标测序或磷蛋白组学实验的数据时,设置自定义背景非常重要。适当的自定义背景将分别包括测序面板中的所有基因或所有已知的磷蛋白。
Stage2B阶段:利用GSEA对序列基因列表进行通路富集分析(步骤6B) Stage 2B:pathway enrichment analysis of a ranked gene list using GSEA(Step 6B)
在GSEA软件(Step 6B) (Box 4)中对一个排序的基因列表进行路径富集分析,GSEA是一种无阈值的方法,它根据所有基因的差异表达等级或其他得分进行分析,不需要事先进行基因筛选。GSEA特别适合,当基因组中的所有或大部分基因(例如RNA-seq数据)都可以使用rank时,推荐使用GSEA。然而,不合适当只有一小部分基因有等级可用时,例如,在一个识别显著突变的癌症基因的实验中(2A阶段;步骤6)。
GSEA搜索的是基因富集在排名基因列表的顶端或底部的路径,其富集程度超出了单凭偶然因素的预期。例如,如果细胞周期中涉及到最高差异表达的基因,这表明细胞周期通路在实验中受到调控。相比之下,如果细胞周期基因在整个排列列表中随机分布,则细胞周期通路可能不会受到显著调控。为了计算一个通路的富集分数(ES), GSEA逐步检查从排名列表的顶部到底部的基因,如果一个基因是通路的一部分,则增加ES,否则降低分数。这些运行和的值被加权,因此在最顶端(和最底部)的序列基因中的富集被放大,而在更中等的序列基因中的富集则没有被放大。ES分数计算为运行和的最大值,并相对于通路大小进行归一化,得到反映列表中通路富集程度的归一化富集分数(NES)。正的和负的NES值分别表示列表顶部和底部的富集。最后,基于排列的P值计算和修正为多个测试基于排列产生错误发现率(FDR)Q值,范围从0(非常重要)到1(不重要)(Box3)。同样的执行分析排名从底部的基因列表来确定富集在底部的列表中的通路。使用FDR Q值阈值(如Q < 0.05)选择得到的通路,使用NES进行排序。此外,GSEA分析的“前沿”方面确定了对检测到的通路富集信号贡献最大的特定基因。
GSEA测定ES统计显著性(P值)的方法有两种:基因组排列和表型排列。
基因集排列检测需要一个排序列表,GSEA将观察到的通路ES与随机取样的匹配大小的基因集(例如,1000次)重复分析得到的分数分布进行比较。
表型排列检测需要所有样本的表达数据(例如,生物复制),以及被称为“表型”的样本组的定义,这些样本组彼此进行比较(例如,病例与对照;肿瘤与正常样本对照)。将观察到的pathway ES与将样本随机打乱表型类别,并重复分析(如1000次)得到的分数分布进行比较,包括对排名基因列表和结果通路ES的计算。
基因组排列推荐用于有限变异性和生物复制的研究。(如:每种情况2到5个)。在这种情况下,差异基因表达值应该在GSEA之外计算,使用的方法包括方差稳定(如edgeR, DESeq和Limma/voom并在通路分析前导入GSEA软件。表型排列应该与更多的复制一起使用(例如,每个条件至少10个)。表型排列方法的主要优点是,相对于基因组排列方法,它在排列过程中保持了具有重要生物学基因相关性的基因组结构。
本方案只涵盖基因组排列,因为它适用于最常见的通路富集分析用例。表现型置换在计算上是昂贵的,而且,对于当前版本的GSEA,需要定制编程来分别计算ESs和差分表达式统计量,以实现数千种表现型随机化。对于高级用户,我们为这个过程提供了一个补充方案(补充方案4)
Stage3:通路富集分析结果可视化与解释(步骤7-13) Stage 3:visualization and interpretation of pathway enrichment analysis results(Steps 7-13)
通路信息本质上是冗余的,因为基因经常参与多个通路,数据库可能通过包含具有许多共享基因的一般和特定通路(如“细胞周期”和“细胞周期的m期”)来分层组织通路。因此,通路富集分析常常强调同一通路的多个版本。将冗通路径折叠成一个单一的生物学主题可以简化解释。我们建议使用EnrichmentMap、ClueGO和其他等可视化方法来解决这种冗余。“Enrichment”是表示富集通路之间重叠的网络可视化(fig 1),而“富集图”是指用Cytoscape创建可视化的应用程序。如果路径共享许多基因,则路径显示为与线(边)相连的圆圈(节点)。节点由ES着色,边缘的大小取决于连接通路共享的基因数量。网络布局和聚类算法自动将相似的路径分组成主要的生物主题。mentmap软件将包含pathway富集分析结果的文本文件和包含原始富集分析中使用的pathway基因集的文本文件作为输入。交互探索通路ES(过滤节点)和通路之间的连接(过滤边缘)是可能的(步骤9A(xii和xiii)和9B(xiii和xiv))。多个富集分析结果可以同时显示在一个富集图中,在这种情况下,每个富集点使用不同的颜色。如果选择加载基因表达数据,点击通路节点,将显示通路中所有基因的基因表达热图。
富集图有助于识别有趣的路径和主题。首先,应确定预期的主题,以帮助验证通路富集分析结果(阳性对照)。例如,生长相关通路和癌症的其他特征有望在癌症基因组数据集的分析中被识别出来。第二,以前与实验环境无关的通路作为潜在的发现被更仔细地评估。首先应该研究ESs最强的通路和主题,然后逐步减弱信号(步骤12)。第三,更详细地检查有趣的通路,检查通路内的基因(例如,表达热图和GSEA前沿基因)。此外,如果可以的话,可以使用PathVisio等工具,在pathway Commons、Reactome、KEGG或WikiPathways等数据库的通路图上覆盖基因表达值。如果没有可用的图表,可以使用STRING或GeneMANIA等工具与Cytoscape一起定义用于表达覆盖的通路基因之间的相互作用网络。这有助于在视觉上识别实验中变化最大的通路成分(如单基因或整个信号级联)(如差异表达)。此外,可以通过整合基因组使用富集图后分析工具对miRNA或转录因子进行目标检测来寻找富集途径的主调控因子.最后,可以发表通路富集分析结果来支持科学结论(例如,两种癌症亚型的功能差异),或者用于假设生成或实验计划,以支持新通路的识别。此网站(http://www.pathwa ycommons.org/guide/)提供了更多的途径富集分析示例和对核心概念的更深入解释。
优势和局限 Advantages and limitations
与单基因、转录本或蛋白质分析相比,组学数据的通路富集分析有几个优点。
首先,它以两种方式提高了统计能力:(i)它汇集了与给定细胞机制相关的所有基因和基因组区域的突变计数,提供了更多的计数,这使得统计分析更加可靠;(ii)它将维度从数以万计的基因或数以百万计的基因组区域(如SNPs)减少到数量少得多的“系统”或“通路”,从而降低了多重假设检验的成本。其次,结果往往更容易解释,因为分析是在“细胞周期”等熟悉概念的层次上进行的。第三,该方法可以帮助确定潜在的致病机制和药物靶点。第四,从相关但不同的数据中获得的结果可能更具可比性,因为结果被投影到更小的共享特征空间(如,有限数量的通路);第五,该方法有助于整合不同的数据类型,如基因组学、转录组学和蛋白质组学,这些数据类型都可以映射到相同的路径。因此,将疾病数据投射到已知的机制上增加了统计和解释能力。
在解释通路富集分析结果时,通常包括本方案所涵盖的结果时,需要考虑以下限制。根据组学数据类型(参见“应用于不同组学数据”一节),还存在其他限制。具体和可选的通路富集分析方法的优缺点在“与替代方法的比较”一节中介绍。
●富集分析对于多个基因具有强生物信号(如差异表达)的通路更有效。例如,在转录组学实验中,我们假设进化已经优化了一个细胞,只有在需要的时候才表达一个通路,而这个通路的激活或失活可以被识别为一个通路中许多基因的协调活动。活性仅受少数基因控制或不受基因表达(如翻译后调控)控制的通路将永远不会被视为丰富的。一些通路分析方法通过激活和抑制基因相互作用来构建通路活性的定量模型,其中包括未差异表达但仍是重要调控因子的基因。然而,这些方法需要具有详细生化和调控基因的通路模型。
●通路边界往往是任意的,不同的数据库会对特定通路涉及哪些基因存在分歧。通过使用多个数据库,可以分析多个路径定义,其中一些可能比另一些更能解释实验数据。
●一些途径富集方法,如基于Fisher 's精确检验的方法,在统计上更有可能识别出更大的途径。用户可以通过选择分析中考虑的基因集大小的上限来解决这个限制。
●在基因列表中排名较高的多功能基因可能导致许多不同途径的富集,其中一些途径与实验无关。排除这些基因后重复分析,可能会发现富集过度依赖于其存在的通路,或证实通路富集的稳健性。
●通路数据库,因此富集结果偏向于已知的通路。事实上,通路富集分析忽略了没有通路注释的基因,有时被称为“基因组的暗物质”,这些基因应该单独研究。例如,非编码RNA基因目前缺乏系统的注释,不能直接用于通路富集分析
●大多数富集分析方法对基因之间以及通路之间的统计独立性做出不切实际的假设。有些基因可能总是共同表达的(例如,蛋白质复合体中的基因),而有些通路有共同的基因。因此,标准的FDRs(假定测试之间具有统计独立性)往往比理想情况更保守或更不保守。尽管如此,它们仍然应该被用于调整多个测试,并为探索性分析和假设生排序富集通路。自定义排列测试可能导致对错误发现的更好估计(参见“与其他方法的比较”一节)。
实验设计 Experimental design
仔细的实验设计使通路富集分析受益匪浅。否则,分析可能会揭示由实验偏差或其他混杂因素造成的明显有意义的结果。本节介绍了在执行此方案之前必须考虑的一系列实验因素。
实验条件 Experimental conditions
实验条件必须明确,以便观察到的主要变化是实验者想要监测的反应,并且与感兴趣的生物学问题相关(例如,肿瘤与正常、治疗与未治疗、四种疾病亚型的比较、时间序列)。
数量的复制 Number of replicates
生物复制是从不同的生物体或细胞系中获得的独立处理过的样本,用于测量样本之间的变异性并计算统计显著性(P值)。缺乏复制(即每组一个样本)将不允许鲁棒估计信号的重要性。复制不足可能导致数据中缺乏信号(如无显著差异表达的基因)。在一组样本中,变异越大,就需要更多的生物复制来精确地测量信号。对于变异性较低的系统(即在受控实验室条件下具有相同遗传背景的模型生物,或来自同一克隆的稳定细胞系),每个条件至少推荐三到四个生物复制,用于方差稳定归一化的差异分析。方差稳定使用一个全局统计模型来“稳定”基因方面的方差估计,以减少由少数重复造成的误差。对于变异性较高的实验(如肿瘤样本),需要更多的复制;理想情况下,应使用正式的统计力计算(有时称为敏感性测试)后的先导实验来确定识别差异表达基因或富集途径信号所需的最小重复数。由相同样品的重复实验组成的技术复制通常不需要成熟的实验技术,如RNA-seq,它具有较低的技术变异性,但对新技术有帮助。
混杂因素 Confounding factors
应该避免与实验问题无关的因素之间的差异,或者至少在不同的条件下保持平衡,以便统计技术(如广义线性模型)能够纠正每个因素。常见因素包括测序批次、核酸提取方案、受试者年龄等。否则,可能无法准确地将实验信号从实验响应和混淆因素中分离出来。提前了解重要因素有助于正确的实验设计。统计探索性分析,如聚类分析或主成分分析(PCA),可以帮助识别未知因素。例如,案例和控件应该单独集群,而不是通过处理批处理。
离群值 Outliers
由于主要的实验或技术问题,如污染或样品混淆,离群样本可能与其他样本有很大的不同。或者,它们可能呈现极端的生物学特征,例如具有异常侵略性表型的肿瘤样本。使用PCA或聚类等统计技术可以无偏倚地识别离群样本。通路富集分析应该有或没有异常值,以确保稳健的结果。系统地去除异常值可能有助于减少实验中的变异性。
实验灵敏度 Experimental sensitivity
一些实验方法可以调到更敏感或更不敏感。例如,RNA-seq实验中的读取数影响下游分析。为了量化具有适度变异性的生物系统中的基因表达,并测试具有方差稳定的差异表达,至少需要3到5次重复和1000万个图谱解读。为了研究剪接异构体、检测表达不良的基因或具有复杂细胞混合物(如手术切除标本)的样本,需要更大的测序深度,如5000 - 1亿个映射读图。
通路基因集数据库的选择Choice of pathway gene se database
我们建议在开始时只搜索富集通路基因集,因为这些基因集捕获了熟悉的、容易解释的正常细胞过程。来自Reactome、Panther、HumanCyc和NetPath的GO生物过程术语和人工调控的分子通路是很好的人类路径资源(Box2)
筛选GO通路基因集 Filtering GO pathways gene sets
GO中大量的基因注释来自于自动数据分析,并没有经过人类管理员的验证。这些有证据代码“从电子注释推断”(IEA)。早期的文献告诫人们不要分析和解释IEA标记的注释,而最近的研究表明,这些注释通常和人类管理员所作的注释一样可靠.对于来自人类和常见模型生物体的数据的高可信度分析(这些数据有许多手动管理的注释),我们通常建议比较分析版本(有和没有过滤IEA注释),以验证健壮性。然而,IEA的注释在研究较少的物种中占据了大部分信息,在这些情况下应该默认使用。删除IEA编码的注释可能会使分析偏向于深入研究的生物学过程。
使用非通路基因集 Use of non-pathway gene sets
不同类型的基因组有助于回答各种各样的问题。例如,与microRNA和转录因子靶标相对应的非通路基因集可以用来发现重要的调控因子。然而,同时分析所有可用的基因集类型会降低数据的可解释性。这也可能导致假阴性,因为所进行的试验次数的增加增加了多次试验校正的效果,降低了单个通路的多次试验校正的显著性。因此,我们建议对非途径和途径基因集分别进行分析
基因集大小的考虑 Gene set size considerations
排除大量的小路径往往是有益的,因为它们与较大的路径是冗余的,解释也比较复杂,而且它们的丰富性使得多重测试校正更加严格。大通路也应该被排除,因为这些通路过于普遍(如“新陈代谢”),它们对结果的可解释性没有贡献,当使用某些统计富集方法(如Fisher’s exact test)时,它们的统计意义可能会被夸大。对于分析人类基因表达数据,我们通常建议排除小于 10-15个基因和(虽然在文献中可以找到200 - 2000个基因的上限)大于 200-500个基因。然而,对于非人类生物和其他类型的基因集,可能有不同的基因集大小分布,可能需要包括更大的集。通路的筛选依赖于实验环境,因为不同的生物学领域在通路数据库中有不同的覆盖范围。我们可以通过检测与实验相关的几个感兴趣的通路的大小来确定通路大小的上下界。
使用最新的通路基因集的重要性 Importance of using updated pathway gene sets
通路富集分析依赖于分析中使用的基因集和数据库,近年来许多利用途径富集分析的研究受到过时资源的强烈影响。为了提高研究的重现性和透明度,研究人员应在出版物中报告使用的通路富集分析软件和基因集数据库的分析日期和版本,以及所有分析参数。除了富集图谱,作者还应该考虑添加他们所研究的基因列表和完整的富集通路表作为补充信息。
基因标识符的选择 Choice of gene identifier
基因与许多不同的数据库标识符(id)相关联。们建议使用明确、惟一和稳定的id,因为有些id会随着时间的推移而过时。对于人类基因,我们建议使用Entrez基因数据库id(例如,4193对应MDM2)或基因符号(MDM2是HUGO基因命名委员会推荐的官方符号)。随着基因符号的变化,我们建议同时维护基因符号和Entrez基因id。Profiler和相关的g:Convert工具支持将多个ID类型自动转换为标准ID。
意想不到的通路结果和实验设计 Unexpected pathway results and experimental design
通路分析中所揭示的意想不到的生物学主题可能表明实验设计、数据生成或分析存在问题。例如,细胞凋亡通路的富集可能表明实验方案存在问题,导致在样品制备过程中细胞死亡增加。在这些情况下,在进一步解释数据之前,应仔细审查实验设计和数据生成。
应用于不同的组学数据Applicatin to diverse omics data
该协议使用RNA-seq data7和体细胞突变data6作为示例,因为这些数据类型经常遇到。然而,我们提出的通路富集分析的一般概念适用于许多可以生成基因列表的实验类型,如单细胞转录组学、CNVs、proteomics、phosphphoproteomics、DNA甲基化和metabolomics66。大多数数据类型都需要修改方案,这里只简要讨论一下。对于某些数据类型,需要专门的计算方法来生成适合于通路富集分析的基因列表,而对于其他数据类型,则需要专门的通路富集分析技术。必须考虑特定于数据类型和实验方法的问题,包括:
●对于某些数据类型,建议使用不同的基因标识符。我们推荐蛋白质的UniProt加入数(例如,MDM2的Q00987)和代谢物的人类代谢组数据库id(例如,MDM2的Q00987)。, ATP记为HMDB00538)。
●某些类型的组学实验通过设计只捕获基因或蛋白质的一个子集。为了解决这种有限的覆盖,途径富集分析必须定义一个自定义的背景基因集,该基因集可以在实验中测量。例如,磷酸化蛋白组学实验只测量具有一个或多个磷酸化位点的蛋白,因此必须使用编码磷酸化蛋白的一组基因作为自定义背景基因集。否则,通路富集分析将显示激酶信号传导和蛋白磷酸化等一般过程的P值过高。
●ChIP-seq实验中转录因子结合位点等短非编码基因组区域的通路富集分析需要进一步考虑。基因组区域必须映射到蛋白质编码基因,并纠正偏差,如在较长的基因中增加信号。GREAT67等工具会自动执行这两项任务。
●跨越多个基因的大基因组间隔(例如,来自全基因组关联、CNV和差异甲基化区域)需要专门的富集检测,如PLINK CNV基因集负载测试或INRICH。标准的富集测试常常揭示基因组中聚集的基因,由于每个基因被错误地当作独立信号计算,这些基因的信号在统计上被严重夸大。与基因组位置相关的基因类型包括嗅觉受体、组蛋白、主要组织相容性复合体(MHC)成员和同源框转录因子。一个简单的解决方法是在富集分析之前,从每个功能相同的基因组簇中只选择一个有代表性的基因
●对于罕见的遗传变异,病例对照途径“burden”检测是最合适的途径富集分析方法(参见“与其他方法的比较”一节)。
与其他方法的比较 Comparison to alternative methods
通路富集分析方法
本方案推荐使用g:Profiler和GSEA软件进行通路富集分析。g: profiler使用Fisher精确检验分析基因列表,使用修正的Fisher检验对基因列表进行排序。它通过R和Python编程语言提供图形化web界面和访问。该软件经常更新,基因集数据库可以作为GMT文件下载(http://biit.cs.ut.ee/gprofiler)。GSEA使用基于排列的测试分析排序的基因列表。该软件通常作为桌面应用程序运行(http://software.broadinstitute.org/gsea)。目前存在数百种通路富集分析工具,由于许多工具依赖于outof-date通路数据库,或者与最常用的工具相比缺乏独特的特征;因此,我们不在这里讨论它们。以下是可供选择的自由通路富集分析软件工具。尽管我们的协议中没有涉及这些工具,但我们建议使用以下工具,基于它们的易用性、独特的特性或高级编程特性。
●Enrichr:这是一个基于web的富集分析工具non-ranked基因列表基于确切概率法。它使用方便,具有丰富的交互式报告功能,包括>100个基因集数据库(称为文库),其中>18万个基因集在多个类别。功能类似于本协议中描述的g:Profiler web服务器。
●Camera:这个R Bioconductor包分析基因列表和纠正等inter-gene相关性明显的基因co-expression数据。该软件可作为Bioconductor中的limma包的一部分使用(https://bioconductor.org/packages/release/bioc/html/limma.html;这是一个高级工具,需要编程专家;补充方案3)。
●GOseq:这个R Bioconductor包分析从RNA-seq实验,用户修正如基因长度等协变量选择来基因列表(https://bioconductor.org/packages/release/bioc/html/goseq.html;这是一个高级工具,需要专门的编程知识)
● Genomic Regions Enrichment of Annotations Tool (GREAT):与分析基因列表的常用方法不同,GREAT分析基因组区域,如DNA结合位点,并将其与邻近基因连接,进行通路富集分析(http://bejerano.stanford.edu/great/public/html/)。
可视化工具:
本方案建议使用富集图进行通路富集分析可视化,以帮助解释。EnrichmentMap是一个Cytoscape应用程序,它可以将通路富集分析的结果可视化,并将途径显示为一个网络,其中重叠的途径聚集在一起,以识别结果中的主要生物主题,从而简化解释(http://www.baderlab.org/software/mentmap)。两个有用的可视化工具是:
●ClueGO:这个Cytoscape应用程序在概念上类似于EnrichmentMap,提供了一个基于网络的可视化,以减少路径富集分析结果的冗余。它还包括一个用于使用Fisher 's精确测试分析GO注释的通路富集分析特性。然而,目前它只支持GO基因集。
●PathVisio:这个桌面应用程序提供了一个互补EnrichmentMap和ClueGO可视化方法。PathVisio使用户能够在感兴趣的基因和蛋白质相互作用的背景下直观地解释组学数据。根据用户提供的组学数据(https://www.pathvisio.org), PathVisio颜色通路基因。这是PathVisio相对于mentmap和ClueGO的主要优势。
拓扑感知通路分析方法Topology-aware pathway analysis methods
大多数通路富集分析方法对同一通路中的所有基因都进行统一处理,忽略了基因间的相互作用。相比之下,拓扑感知方法显式地模拟基因间的相互作用。CePa、GANPA和THINK-Back使用物理基因相互作用或共表达网络为每个通路中的每个基因分配权重。权重可以通过测量网络中基因的重要程度,如度、基因连接数、中介中心度等来确定,可以集成到传统的途径富集分析方法中,如GSEA。尽管调控和生化基因相互作用有用且可能更准确,但与物理相互作用网络和共表达相比,调控和生化基因相互作用可用于更少的基因和通路。我们预期这些方法将变得更有用,因为更多的基因相互作用的途径是在详细的分子实验的特点。然而,目前从文献中收集和整理高质量和生物化学详细的通路数据是复杂和昂贵的。因此,在可预见的未来,本方案中描述的通路富集分析方法可能仍将是最广泛使用的方法
未来的角度 Future perspective
目前的通路富集分析方法为基因组学实验中活跃的途径提供了一个有用的高层次概述。然而,这些方法考虑了一个只涉及基因集的简化通路视图。下一代通路分析方法将整合更多的生物通路细节,建立基于多种类型基因组数据的多样本检测的通路模型,并考虑数据中的正调控关系和负调控关系。例如,用单细胞RNA-seq数据参数化的定性数学模型可能有一天能够准确预测能够治疗正在研究的特定疾病的药物组合
方案概述Overview of the protocol
这个循序渐进的方案解释了如何使用g:Profiler(过滤基因列表)和GSEA(未过滤的、全基因组的、排序的基因列表)完成通路富集分析,然后使用富集图进行可视化和解释。为g:Profiler分析提供的示例数据是癌症基因组图谱(TCGA)外显子测序数据中发现的12个类型的3200个肿瘤的频繁体细胞单核苷酸变异(SNVs)基因列表。GSEA分析提供的示例数据是TCGA定义的两种卵巢癌亚型的差异表达基因列表。
材料 Materials
设备Equipment
硬件Hardware
●可上网且内存≥8gb的个人电脑。1gb内存足够运行GSEA分析;然而,Cytoscape(需要运行mentmap软件)需要≥8gb 内存。
软件Software
●使用g:Profiler进行通路富集分析的当代web浏览器(如Chrome)(步骤6)。
●g:Profiler(https://biit.cs.ut.ee/gprofiler/)
●运行GSEA和Cytoscape需要Java标准版(http://www.oracle.com/technetwork/java/javase/downloads/index.html)
●GSEA桌面应用程序(http://software.broadinstitute.org/gsea/downloads.jsp)用于通路富集分析(步骤6B)。
●富集图可视化需要Cytoscape桌面应用程序(http://www.cytoscape.org/download.php)和以下Cytoscape应用程序:mentmap, v.3.1或更高;clusterMaker, v.0.9.5或更高;WordCloud, v.3.1.0或更高;AutoAnnotate,v,1.2.0或更高。通过从Cytoscape应用程序商店安装“EnrichmentMap Pipeline Collection”(http://apps.cytoscape.org/apps/mentmappipelinecollection),可以方便地下载并一起安装这些软件程序。
输入数据Input data
CRITICAL(重点:):
我们提供了可下载的示例文件,这些文件在整个方案中都被引用(补充表1-13)。我们建议在启动之前将所有这些文件保存在个人项目数据文件夹中。我们还建议创建一个额外的结果数据文件夹来保存在执行协议时生成的文件。
●感兴趣的基因列表或排序基因列表
步骤6A的示例数据。g:Profiler需要一个文本文件或电子表格中的每行一个基因列表,准备复制并粘贴到web页面:为此,我们使用TCGA外显子组测序数据中发现的3200个12种类型肿瘤的频繁体细胞SNVs基因。MuSiC癌症驱动突变检测软件被用于查找127癌症驱动基因显示高于预期的基因突变频率在癌症样本(补充表1,来自参考文献6补充表4 的列B。)。基因依据极影的重要性(FDR Q值)和突变频率(没有显示)降序排名。
步骤6B的示例数据。GSEA需要一个带有基因评分的RNK文件。RNK文件是一个两列文本文件,第一列是基因id,第二列是基因得分。基因组中的所有(或大部分)基因都需要有一个分数,而基因id需要匹配GMT文件中使用的那些。我们提供了TCGA中卵巢癌差异表达基因的排序列表(补充表2)。本队列先前根据基因表达数据分为四种分子亚型,分别为分化型、免疫反应型、间质型和增殖型。我们比较了免疫反应性和间充质亚型,以证明该方案。补充方案1的步骤5显示了如何创建该文件。
●通路基因数据库
- 步骤6A, g:Profiler维护来自多个来源的最新的通路基因集集,不需要用户进一步输入,但步骤6B (GSEA)需要通路基因集数据库。补充表3包含一个用于标准GMT格式的通路富集分析的通路基因集数据库,可从http://baderlab.org/GeneSets下载。该文件在2017年7月1日下载包含从8个数据来源的通路:GO、Reactome、Panther、NetPath、NCI79、MSigDB curated gene sets (C2 collection,不包括Reactome和KEGG)、MSigDB Hallmark (H collection)和HumanCyc。可以从http://baderlab.org/GeneSets获得每月更新一次的基因集。GMT文件是一个文本文件,其中每一行代表一个单一通路的基因集。每一行都包含一个通路ID、一个名称和以制表符分隔的格式列出的相关基因。
过程Procedure
软件安装 Software installation 时间5分钟
1.从方案补充资料中下载所需的输入输出文件。
●创建两个目录,项目数据文件夹和结果数据文件夹。
●将下载的所有输入和示例输出文件放入项目数据文件夹。
●在完成方案的过程中,将新生成的文件放入结果数据文件夹。
2 .安装Java 8或更高。请遵循http://www.oracle.com/technetwork/java/javase/downloads/index.html上的Java JRE下载和安装说明
3.下载最新版本的GSEA。我们建议使用javaGSEA桌面应用程序在http://www.broadinstitute.org/gsea/downloads.jsp上。需要免费注册。
4.从http://www.cytoscape.org下载最新版本的Cytoscape。Cytoscape v.3.6.0或更高的要求。
5.所需的Cytoscape应用程序。
●启动Cytoscape。
●进入Apps→App Manager(即,打开Apps菜单,选择item App Manager)。
●在Install Apps选项卡搜索栏中,搜索EnrichmentMap。
●点击中心面板上的EnrichmentMap Pipline Colletion。验证它是v.1.0.0或更高。
●点击Install按钮。
●转到当前已安装选项卡,验证应用程序(EnrichmentMap、clusterMaker2、WordCloud和AutoAnnotate)是否已安装。
通路富集分析 Pathway enrichment analysis 3-20分钟
6.可以使用g:Profiler(选项A)分析数十到数千个基因的平面(未排序的)基因列表。从组学数据编译一个基因列表需要一个统计阈值。相比之下,全基因组基因列表适合使用GSEA进行途径富集分析(选项B)。使用GSEA进行分析的基因列表不需要使用统计阈值进行预先筛选。部分、过滤排序的基因列表也可以用g:Profiler进行分析。选择步骤6A或6B,这取决于您拥有的基因列表的类型。
(A)利用g:Profiler对基因列表进行通路富集分析
(i)打开g:Profiler网站http://biit.cs.ut.ee/gprofiler/ (Fig. 2) 。

(ii) 将基因列表(补充表1)粘贴到屏幕左上角的查询字段中。基因列表可以是空格分隔的,也可以是每行一个。默认情况下,用于分析的生物体是Homo sapiens。输入列表可以包含基因和蛋白质id、符号和登录号的混合。重复的和无法识别的id将自动删除,并且在提交查询后,可以在交互式对话中细化模糊的符号
(iii)勾选“Ordered query”旁边的方格。该选项将输入视为有序的基因列表,并在列表的开头优先排列突变ESs较高的基因。
(iv)(可选)勾选No electronic GO annotations旁边的复选框。此选项将丢弃不太可靠的GO注释(IEAs),这些注释不需要手动检查。
(v)使用右侧菜单设置基因注释数据过滤器。我们建议初始途径富集分析只包括GO生物过程(BPs)和Reactome分子途径。选中这两个复选框并取消选中菜单中的所有其他复选框。
(vi)点击“Show Advanced Options”可设置附加参数.
(vii)将下拉菜单中的功能类别大小设置为5 (' min ')和350 (' max ')。大路径的解释价值有限,而众多的小路径由于过多的多重检验而降低了统计能力
(viii)在下拉菜单中将查询/术语交集的大小设置为3。分析将只考虑在输入基因列表中包含三个或更多基因的更可靠的通路。
(ix)点击 g:Profile!来运行分析。将显示一个图形化的热图图像,沿着y轴(左侧)显示检测到的路径,沿着x轴(顶部)显示输入列表的相关基因。结果的路径被分层组织成相关的组。Pfofiler默认使用图形输出,当发现大量路径时,切换到文本输出。g:Profiler只返回具有统计意义的路径,P值经过多次测试校正(称为Q值)。默认情况下,报告Q值<0.05的结果。g:Profiler报告无法识别和模糊的基因id,可以手动解析.
(x)使用下拉菜单输出类型并选择选项Generic Enrichment Map(选项卡)。该文件是需要可视化的路径结果用Cytoscape 和 EnrichmenMap
(xi) 再次使用Profile 更新的参数运行分析。所需的链接下载数据(GEM)格式将出现在g:Profiler接口下。从链接下载文件并将其保存在步骤1中创建的结果数据文件夹中。示例结果见补充表4.
(xii)点击选项表格底部的Advanced Opations链接名称,下载所需的GMT文件。GMT文件是一个压缩的ZIP归档文件,包含g:Profiler(例如,gprofiler_hsapiens. name .gm . ZIP)使用的所有基因集。基因集文件按数据源划分。下载并解压ZIP存档到项目文件夹。该分析所需的所有基因集都在hsapiens.path . Name.gmt(Supplementary_Table5_hsapiens.pathways.NAME.gmt)。将保存的文件放在步骤1中创建的结果数据文件夹中。
(B)利用GSEA对序列基因表进行通路富集分析 时间~ 20分钟
(i)打开下载的GSEA文件(GSEA .jnlp),启动GSEA(fig3)。
(ii)点击GSEA分析部分步骤左上角的Load Data
(iii)在Load Data选项卡中,单击Browse for files…
(iv)找到项目数据文件夹,并选择supplementary_table2_mesenvsimmuno_rnaseq_rank.rnk文件。还可以使用shift-click(补充表3)等多重选择方法选择pathway gene set definition (GMT)文件,点击Choose按钮继续。消息框指示文件已成功加载。单击OK按钮继续。
CRITICAL STEP (重要一步)GSEA还提供了自己的基因集文件,这些文件可以通过GSEA接口从MSigDB resource直接访问。这些文件不需要导入GSEA。要定义GMT文件,请在Select one或more genesets对话框的第一个选项卡gene Matrix(来自网站)中找到MSigDB基因集文件。MSigDB基因集文件的最新版本以粗体显示,但是也可以访问早期版本。要选择多个基因集文件,在Windows中按住control键或在macOS中按住command键时单击所需的文件。
(v)点击工具栏下的Run gseapreanked。一个预先排序的基因列表选项卡上的Run GSEA将出现。

下列参数的说明:
(vi)基因集数据库。点击右边的“(…)”按钮,等待几秒钟,基因集选择窗口就会出现。使用右上角的箭头进入Gene matrix (local gmx/gmt)选项卡。单击下载的本地GMT文件“Supplementary_Table3_Human_GOBP_AllPathways_no_GO_iea_July_01_2017_symbol”。,然后单击窗口底部的OK。
(vii)排列数。这指定了随机化基因集以创建空分布以计算P值和FDR Q值的次数。使用1,000个排列的默认值。
CRITICAL STEP:排列次数越多,计算时间越长。为了计算每个基因集的FDR Q值,通过遍历每个基因集中的基因并重新计算随机集的P值来随机化数据集。这个参数指定了随机化操作的次数。执行的随机化越多,FDR Q值估计就越精确(到一定程度,因为最终FDR Q值将稳定在实际值)。在具有16 G 内存和i7 3.4 ghz处理器的Windows机器上,使用上述定义的参数对我们的示例集进行10、100、500或1000个随机化分析,分别需要155秒、224秒、544秒和1012秒。
(vii)排序列表。通过单击最右边的箭头并选择排序基因,显示已排序的文件(补充表2)
(ix)点击基本字段旁边的“Show”按钮,以显示其他选项.
(x)分析的名字。将默认的“my_analysis”更改为一个特定的名称,例如,“Mesen_vs_Immuno”
(xi)最大尺寸:排除较大的集。默认情况下,GSEA将上限设置为500。将这个设置为200从分析中删除较大的集合。
(xii)将结果保存在此文件夹中。导航到GSEA应该保存结果的文件夹。我们建议您选择步骤1中创建的结果数据文件夹。否则,GSEA将使用主目录中的默认位置' gsea_home/output/[date] '。
Running GSEA
(xiii)点击窗口底部的Run按钮运行GSEA。如果按钮不可见,则展开窗口。窗口左下角的GSEA reports窗格将显示“正在运行”状态。完成后将更新为“Success”。这将是一个长时间运行的过程,具体取决于计算机的速度.
Examination of GSEA results GSEA结果检查
(xiv) GSEA分析完成后,屏幕左下角会出现一个绿色通知“成功”。所有GSEA输出文件将自动保存,并可在GseaPreranked接口(步骤6B(xii))中指定的文件夹中使用。单击Success在web浏览器中打开结果。这些通路富含top-ranking基因(如,上调)即在第一个集合中显示(' na_pos ';本方案中“皮质间的”)和富含bottome-ranked基因的通路。(如,下调)显示在第二组(' na_neg ';免疫反应性的)(fi'g4)
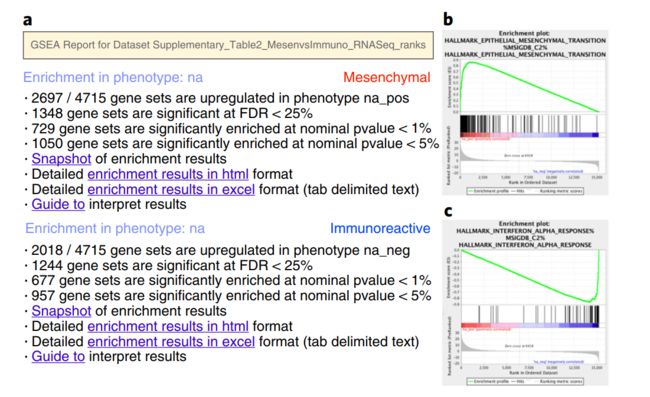
(xv)在“网页浏览器结果摘要”内,按一下“结果”下的“快照”连结,可浏览前20项结果。第一个表型(' na_pos ')最重要的通路应该在最顶端(即(图左侧)。相反,对于第二种表型(' na_neg '),最重要的通路应该清楚地显示富集在底端(即,基因下调(图右侧)(fig4)
CRITICAL STEP:使用表达式数据作为输入(而不是预先计算的秩文件)、表现型标签(即提供生物条件或样本类)作为每个样本的输入,并在GSEA ' cls '文件中指定。运行GSEA时,指定了两种表型进行比较以进行差异基因表达分析,这些表型用于通路富集结果文件。相比之下,在GSEA预先排序的分析中(即排名,当一个基因列表由用户提供),GSEA自动标签一个表型na_pos”(对应于浓缩在基因排名列表的顶部,在那里“na”意味着表型标签是“不可用”)和其他“na_neg”(对应于富集基因底部的排名列表)。EnrichmentMap软件也使用这个约定,指定第一个表现型为“阳性”,第二个表现型为“阴性”。
(xvi)在web浏览器结果摘要中,单击HTML格式的详细富集结果,并使用行号检查FDR Q值<0.05的路径数量,以确定协议下一步富集map的适当阈值。如果在Q < 0.05没有通路的报道,可以使用更宽松的阈值,如Q < 0.1或Q < 0.25(fig5)。阈值Q < 0.25提供了非常宽松的过滤,在这个水平上发现数千个富集的通路并不罕见。稳健分析应该使用Q < 0.05或更低的截止值。仅使用未纠正的P值进行过滤是不合适的,也不推荐使用.
