1.TCGA数据库 patient_barcode--那些需要了解的“暗号”
- 在TCGA数据库中,相同的参与者可能既有肿瘤组织也有正常组织的数据,生存分析时需要将肿瘤数据选出来,进行下一步分析;如果是做差异分析,则可能可能是比较肿瘤和正常组织;
- 详细的可以看概况和细节

- 最近用得较多的是,通过Sample中的数字部分来区分tumor(<10)和normal(>=10)
| 编码 | 代表内容 | 缩写 |
|---|---|---|
| 1 | Primary Solid Tumor | TP |
| 2 | Recurrent Solid Tumor | TR |
| 3 | Primary Blood Derived Cancer - Peripheral Blood | TB |
| 4 | Recurrent Blood Derived Cancer - Bone Marrow | TRBM |
| 5 | Additional - New Primary | TAP |
| 6 | Metastatic | TM |
| 7 | Additional Metastatic | TAM |
| 8 | Human Tumor Original Cells | THOC |
| 9 | Primary Blood Derived Cancer - Bone Marrow | TBM |
| 10 | Blood Derived Normal | NB |
| 11 | Solid Tissue Normal | NT |
| 12 | Buccal Cell Normal | NBC |
| 13 | EBV Immortalized Normal | NEBV |
| 14 | Bone Marrow Normal | NBM |
| 15 | sample type 15 | 15SH |
| 16 | sample type 16 | 16SH |
| 20 | Control Analyte | CELLC |
| 40 | Recurrent Blood Derived Cancer - Peripheral Blood | TRB |
| 50 | Cell Lines | CELL |
| 60 | Primary Xenograft Tissue | XP |
| 61 | Cell Line Derived Xenograft Tissue | XCL |
| 99 | sample type 99 | 99SH |
2.TCGA数据下载
- 数据下载
library(TCGAbiolinks)
query <- GDCquery(project = "TCGA-BRCA",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts",
barcode = clin[1:500,1])
GDCdownload(query)
BRC_DATA1<-GDCprepare(query,save=FALSE)
library(SummarizedExperiment)
BRC_DATA1_1 <- assay(BRC_DATA1)
- workflow.type有多个选择,HTSeq - Counts,HTSeq - FPKM-UQ,HTSeq - FPKM等;
- GDCquery设置好要下载的内容;
- GDCdownload进行下载;如果已经下载过了,运行该步骤会有数据已经下载过的提示;
- GDCprepare将下载的数据(文件夹套文件夹的格式)进行整理,得到一个表格;但这次最后得到的是一个SummarizedExperiment objects,用
SummarizedExperiment中的assay将内容提取出来即可; - 临床数据下载及药物信息
library(TCGAbiolinks)
clinic <- GDCquery_clinic(project = "TCGA-BRCA",
type = 'Clinical')
####限定查药物信息时才能找到更具体的信息
query <- GDCquery(project = "TCGA-BRCA",
data.category = "Clinical",
file.type = "xml")
GDCdownload(query)
clinical <- GDCprepare_clinic(query, clinical.info = "drug")
- 个性化的
TNBC分型
TCGA中TNBC的数据是基于Her2、ER、PR的IHC结果来进行划分的,相应的表型数据是这样获得的:内容整理自TNBCsub
1.点击链接[GDC Data],选择界面右下角Legacy Archive(https://portal.gdc.cancer.gov/)

2.Primary Site和Project选择;

3.Files中,Data Type选择Clinical data,Data Format选择Biotab,右侧文件选择
nationwidechildrens.org_clinical_patient_brca.txt;

3.KMplot
a.看下这个图是用了什么数据?
b.看下这些数据是怎么来的?
c.如何区分是否有差异?log-rank test
- 一系列代码
library(survival)
library(survminer)
fit.surv1 <-Surv(tumor_clin$time,as.numeric(tumor_clin$Status))
fit<-survfit(fit.surv1~group,data=voom_group)
summary(fit)
ggsurvplot(fit)
tumor_clin$time生存分析中的时间;
tumor_clin$Status生存分析中的Alive-0和Dead-1;
group对应的数据中的分组信息的列的列名;
summary(fit)查看数据

- a中提到的图,即是下图,其实KMplot主要的目的即是生存曲线可视化
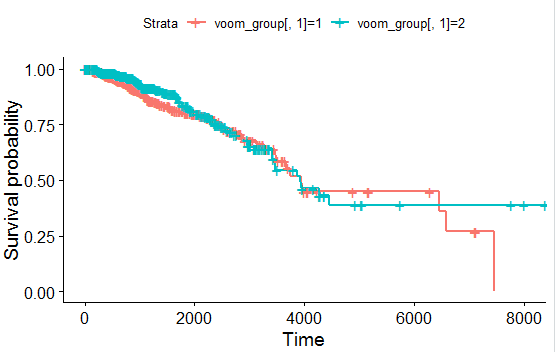
- 演示survival的计算(计算了一组):
clin_group<- tumor_clin[,c('Status','time')]
group <- voom_group[,1]
#####取出对应分组的临床信息
group_1 <- clin_group[group==1,]
group_2 <- clin_group[group==2,]
#####取出对应分组的对应Status(Alive|Death)信息
group_1_A<- group_1[group_1$Status==0,]
group_1_D<- group_1[group_1$Status==1,]
group_2_A<- group_2[group_2$Status==0,]
group_2_D<- group_2[group_2$Status==1,]
#####对1组下的Status信息按照time进行排序
A_1<- group_1_A[order(group_1_A$time),]
D_1<- group_1_D[order(group_1_D$time),]
######将相同time的event的计数进行加和
D_1<- data.frame(time = as.numeric(D_1$time[!duplicated(D_1$time)]),
event= as.numeric(by(D_1,D_1$time, function(x){length(x[,1])})))
#####将相同time的censor(alive)的计数进行加和
A_1<- data.frame(time = as.numeric(A_1$time[!duplicated(A_1$time)]),
censor= as.numeric(by(A_1,A_1$time, function(x){length(x[,1])})))
#####这一组的总人数-event(death)-censor(alive|依据时间排序后,`此death对应时间`之前的alive)
y<- function(x){nrow(group_1)-sum(D_1$event[D_1$time- 至此所求为1组的数据,2组数据同理;
- KMplot涉及到的数据很少,Status、time和分组信息(依据处理或表达量等进行分组)等等,Status中一般Alive和Death用0,1代表或1,2代表(记住就行,别问为什么);
> D_1
time event n.risk step.survival surv
1 26 1 519 0.9980732 0.9980732
2 116 1 501 0.9980040 0.9960811
3 158 1 500 0.9980000 0.9940889
4 160 1 499 0.9979960 0.9920967
5 172 1 496 0.9979839 0.9900965
6 174 1 494 0.9979757 0.9880923
7 302 1 480 0.9979167 0.9860338
8 320 1 473 0.9978858 0.9839491
9 322 1 471 0.9978769 0.9818601
10 336 1 467 0.9978587 0.9797576
11 348 1 463 0.9978402 0.9776415
12 365 1 458 0.9978166 0.9755069
13 377 1 447 0.9977629 0.9733245
14 385 2 441 0.9954649 0.9689104
15 426 1 423 0.9976359 0.9666198
16 446 1 412 0.9975728 0.9642736
17 524 1 372 0.9973118 0.9616815
18 538 1 366 0.9972678 0.9590540
19 558 1 355 0.9971831 0.9563524
20 571 1 351 0.9971510 0.9536278
21 584 1 344 0.9970930 0.9508556
22 612 1 332 0.9969880 0.9479916
23 614 1 331 0.9969789 0.9451275
#####未展示所有,画图用的是time和surv这两列
4.log-rank检验
- log-rank检验是比较生存曲线是否有差异的最常用的方法,非参数检验。
- 零假设是两组之间的生存率没有差异。
- log-rank统计量大致满足为卡方分布。
fit.surv1 <-Surv(tumor_clin$time,as.numeric(tumor_clin$Status))
log_rank_p <- apply(tumor_voom, 1, function(values1){
group=ifelse(values1>median(values1),2,1)
data.survdiff=survdiff(fit.surv1~group)
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1)
return( p.val)
})
- 这里是批量log-rank检验的代码;
- 用survdiff函数的
$chisq、pchisq函数、df=length(data.survdiff$n) - 1对p值进行计算(上面有提到,大致满足卡方分布); - survdiff的结果返回如下:
Oberved是观察到的event的个数;Expected是理论的event的个数;(O-E)^2/E即直观反应到的计算结果;V用data.survdiff$var可以得到(还没细研究);(O-E)^2/V即构建的log-rank的统计量,满足卡方分布的是它;
> data.survdiff
Call:
survdiff(formula = fit.surv1 ~ group, data = voom_group)
N Observed Expected (O-E)^2/E (O-E)^2/V
group=1 545 79 72.1 0.656 1.25
group=2 545 73 79.9 0.592 1.25
Chisq= 1.3 on 1 degrees of freedom, p= 0.3
5.coxph(Cox proportional hazards)
- 用于描述不同变量对于生存的影响;
- 该方法不对“生存模型”做出假设,假设变量对生存的影响随时间变化是恒定的,并且在一个尺度中具有累加效应,因此不是真正的非参数,为半参数;生存曲线可视化无交叉表示满足PH设定;
- 单因素cox批量的代码:
fit.surv1 <-Surv(tumor_clin$time,as.numeric(tumor_clin$Status))
cox_p <- apply(tumor_voom, 1, function(values1){
group=ifelse(values1>median(values1),2,1)
fit <- coxph(fit.surv1~group)
fit1 <- exp(fit$coefficients)
p.val <- summary(fit)$logtest["pvalue"]
result <- list(fit1,p.val)
return(result)
})
- 分组时,将组别用数字2和1来代替,比如,根据表达量高低进行分组,看某基因高表达是风险增加还是降低;所给出的HR即使
组2相对组1的值; - 结果格式如下:
> summary(coxph)
Call:
coxph(formula = fit.surv1 ~ group, data = voom_group)
n= 1090, number of events= 152
coef exp(coef) se(coef) z Pr(>|z|)
group -0.1818 0.8338 0.1627 -1.117 0.264
exp(coef) exp(-coef) lower .95 upper .95
group 0.8338 1.199 0.6061 1.147
Concordance= 0.546 (se = 0.024 )
Likelihood ratio test= 1.25 on 1 df, p=0.3
Wald test = 1.25 on 1 df, p=0.3
Score (logrank) test = 1.25 on 1 df, p=0.3
-
exp(coef)即HR,此处为0.8338;
HR = 1: 无风险
HR < 1: 风险降低
HR > 1: 风险增加
-
lower .95和upper .95为HR的95%CI的下限和上限;可以通过$conf.int[3]和$conf.int[4]调出来; - Likelihood ratio test、wald test和Score(logrank)test的检验结果均有展示;
【参考内容】
1.各种格式TCGA数据下载
2.cox-survival
3.survival rate计算
课程分享
生信技能树全球公益巡讲
(https://mp.weixin.qq.com/s/E9ykuIbc-2Ja9HOY0bn_6g)
B站公益74小时生信工程师教学视频合辑
(https://mp.weixin.qq.com/s/IyFK7l_WBAiUgqQi8O7Hxw)
招学徒:
(https://mp.weixin.qq.com/s/KgbilzXnFjbKKunuw7NVfw)