使用Python对fasta格式的序列进行基本信息统计
预期设计输出文件中包括fasta文件名,序列长度,GC含量以及ATCG各自的含量。
Python脚本编辑
使用的文件
test.fasta
stat.py
输入 sys模块
#!/usr/bin/env python
import sys
从命令行获得文件名称
file_fasta = sys.argv[1]
#获得文件名
file_name = file_fasta.split('.')
name = file_name[0]
sys.argv[1]模块是从程序外部获取参数的桥梁,可以将命令行的参数输入到py程序内。
sys.argv[0]是程序本身,sys.argv[1]是程序后跟着第一个参数。
我们将文件名作为输入参数,这一步在最后运行有展示。
在结束输出时会输出一个包含统计信息的txt文件,我们将用fasta文件名作为txt文件的前缀,所以我们需要获取fasta文件的名字。
.split('.')是将file_fasta以.为分隔符分开会生成'test','txt',赋值给file_name则file_name会包含着两个字符。
file_name[0]则是取第一个值'test',python中默认第一个数字是0,所以不能输入1。
进行序列信息统计的函数
序列的长度很好统计使用len函数即可,但是GC含量和ACTG的百分比计算需要费点事情。
使用def制作一个函数
Python 使用def 开始函数定义,紧接着是函数名,括号内部为函数的参数,内部为函数的 具体功能实现代码
def get_info(chr):
chr = chr.upper()
count_g = chr.count('G')
count_c = chr.count('C')
count_a = chr.count('A')
count_t = chr.count('T')
命名这个函数为get_info,内部参数为chr
在咱们会将fasta中ATCG的碱基内容赋值给chr,碱基可能有大写有小写,所以我们使用.upper将所以字符变成大写。
再使用.count('G')统计ATCG各自的数量并赋值给对应count_g,我们用ATCG各自的统计数可以在后面计算中免疫N值干扰。
gc = (count_g + count_c) / (count_a + count_t + count_c + count_g)
A = (count_a) / (count_a + count_t + count_c + count_g)
T = (count_t) / (count_a + count_t + count_c + count_g)
C = (count_c) / (count_a + count_t + count_c + count_g)
G = (count_g) / (count_a + count_t + count_c + count_g)
gc_con = '{:.2%}'.format(gc)
A_content = '{:.2%}'.format(A)
T_content = '{:.2%}'.format(T)
C_content = '{:.2%}'.format(C)
G_content = '{:.2%}'.format(G)
return (gc_con,A_content,T_content,C_content,G_content)
gc含量计算其等于(G的数量+C的数量)/(A的数量+T的数量+C的数量+G的数量)
A的含量等于(A的数量)/(A的数量+T的数量+C的数量+G的数量),其他值的计算以此类推。
.format使用:
"{1} {0} {1}".format("hello", "world")设置指定位置。
'world hello world'
{:.2f} 保留小数点后两位
最后,使用return返回函数结果(gc_con,A_content,T_content,C_content,G_content)
进行函数计算
#进行函数计算
with open(file_fasta,'r') as read_fa:
读取文件内容赋值给read_fa
python中有两个方式打开文件一种是直接使用open("test.fasta","r"),执行完以后f.close()关闭。
注释:"r"只读模式打开文件;"w"以只写模式打开文件,这种模式下输入内容会覆盖原有内容;"a"以追加模式打开一个文件,这个模式会把新内容追加到原有内容的末尾,不会覆盖。
这里使用的是第二方式with内置函数,它可以将文件自动关闭。
for val in read_fa:
val = val.strip()
if not val.startswith(">"):
seq_info = get_info(val)
len_fasta = len(val)
将read_fa内容赋值给val。
strip() 方法用于移除字符串头尾指定的字符(默认为空格或换行符),这里使用默认。
然后使用startswith() 方法用于检查字符串是否是以指定子字符串开头,在当不是>开头的行时候,才对核酸序列才进行信息统计。
len() 方法返回字符长度获得片段长度
结果屏幕展示
#结果屏幕展示
print('******\n{0}\nlength:{1}\ngc content :{2}\nA content :{3}\nT content :{4}\nC content :{5}\nG content :{6}\n******'.format(name,len_fasta,seq_info[0],seq_info[1],seq_info[2],seq_info[3],seq_info[4]))
使用\n进行换行,用.format指定值输出位置。
结果输出文件
os.write(fd, str)
write() 方法用于写入字符串到文件描述符 fd
#结果输出文件
file_output = open("{}sum.txt".format(name),'a')
file_output.write('******\n')
file_output.write('{}\n'.format(name))
file_output.write('length:{:d}\n'.format(len_fasta))
file_output.write('gc content :{}\n'.format(seq_info[0]))
file_output.write('A content :{}\n'.format(seq_info[1]))
file_output.write('T content :{}\n'.format(seq_info[2]))
file_output.write('C content :{}\n'.format(seq_info[3]))
file_output.write('G content :{}\n'.format(seq_info[4]))
file_output.write('******')
file_output.close()
脚本运行
执行脚本(linux系统)
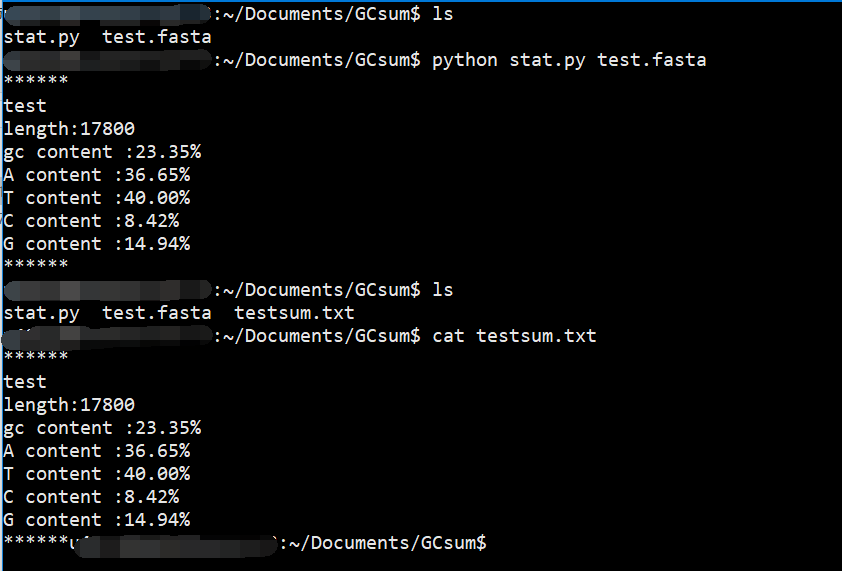
使用ls命令可以看到当前目录下有已经写好的py文件以及数据test.fasta。
运行时注意我们编写时设置从命令行获得文件名称,所以要在后面跟上fasta文件,这样才能成功运行。
运行结束后可以看见屏幕上有结果的打印,同时也生成了testsum.txt。
使用cat命令查看可以看到结果。