BMC Systems Biology

Background
Characterization of drug-protein interaction networks with biological features has recently become challenging in recent pharmaceutical science toward a better understanding of polypharmacology.
Results
We present a novel method for systematic analyses of the underlying features characteristic of drug-protein interaction networks, which we call “drug-protein interaction signatures” from the integration of large-scale heterogeneous data of drugs and proteins. We develop a new efficient algorithm for extracting informative drug-protein interaction signatures from the integration of large-scale heterogeneous data of drugs and proteins, which is made possible by space-efficient representations for fingerprints of drug-protein pairs and sparsity-induced classifiers.
Conclusions
Our method infers a set of drug-protein interaction signatures consisting of the associations between drug chemical substructures, adverse drug reactions, protein domains, biological pathways, and pathway modules. We argue the these signatures are biologically meaningful and useful for predicting unknown drug-protein interactions and are expected to contribute to rational drug design.
Brief:根据药物的化学结构,蛋白质结构及蛋白相关通路等信息来提取潜在的药物与蛋白相互作用

Background
The topological landscape of gene interaction networks provides a rich source of information for inferring functional patterns of genes or proteins. However, it is still a challenging task to aggregate heterogeneous biological information such as gene expression and gene interactions to achieve more accurate inference for prediction and discovery of new gene interactions. In particular, how to generate a unified vector representation to integrate diverse input data is a key challenge addressed here.
Results
We propose a scalable and robust deep learning framework to learn embedded representations to unify known gene interactions and gene expression for gene interaction predictions. These low- dimensional embeddings derive deeper insights into the structure of rapidly accumulating and diverse gene interaction networks and greatly simplify downstream modeling. We compare the predictive power of our deep embeddings to the strong baselines. The results suggest that our deep embeddings achieve significantly more accurate predictions. Moreover, a set of novel gene interaction predictions are validated by up-to-date literature-based database entries.
Conclusions
The proposed model demonstrates the importance of integrating heterogeneous information about genes for gene network inference. GNE is freely available under the GNU General Public License and can be downloaded from GitHub (https://github.com/kckishan/GNE).
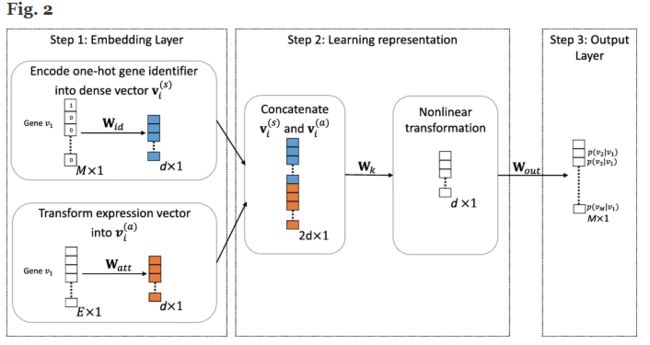
Brief:基因对互作推测,以guilt by association为假设,通过将基因拓扑结构等高维数据嵌入低维空间的空间距离判断基因间相关性,还结合了高通量测序的数据使得有相同特征但没有类似拓扑节后的基因也投影在较近的低维空间中

Background
Systematic fusion of multiple data sources for Gene Regulatory Networks (GRN) inference remains a key challenge in systems biology. We incorporate information from protein-protein interaction networks (PPIN) into the process of GRN inference from gene expression (GE) data. However, existing PPIN remain sparse and transitive protein interactions can help predict missing protein interactions. We therefore propose a systematic probabilistic framework on fusing GE data and transitive protein interaction data to coherently build GRN.
Results
We use a Gaussian Mixture Model (GMM) to soft-cluster GE data, allowing overlapping cluster memberships. Next, a heuristic method is proposed to extend sparse PPIN by incorporating transitive linkages. We then propose a novel way to score extended protein interactions by combining topological properties of PPIN and correlations of GE. Following this, GE data and extended PPIN are fused using a Gaussian Hidden Markov Model (GHMM) in order to identify gene regulatory pathways and refine interaction scores that are then used to constrain the GRN structure. We employ a Bayesian Gaussian Mixture (BGM) model to refine the GRN derived from GE data by using the structural priors derived from GHMM. Experiments on real yeast regulatory networks demonstrate both the feasibility of the extended PPIN in predicting transitive protein interactions and its effectiveness on improving the coverage and accuracy the proposed method of fusing PPIN and GE to build GRN.
Conclusions
The GE and PPIN fusion model outperforms both the state-of-the-art single data source models (CLR, GENIE3, TIGRESS) as well as existing fusion models under various constraints.
Brief:蛋白调控网络推测,通过高斯混合模型

Brief:大型流式数据计算

Background
Anti-tumor necrosis factor alpha (TNF- α) therapy has made a significant impact on treating psoriasis. Despite these agents being designed to block TNF- α activity, their mechanism of action in the remission of psoriasis is still not fully understood at the molecular level.
Results
To better understand the molecular mechanisms of Anti-TNF- α therapy, we analysed the global gene expression profile (using mRNA microarray) in peripheral blood mononuclear cells (PBMCs) that were collected from 6 psoriasis patients before and 12 weeks after the treatment of etanercept. First, we identified 176 differentially expressed genes (DEGs) before and after treatment by using paired t-test. Then, we constructed the gene co-expression modules by weighted correlation network analysis (WGCNA), and 22 co-expression modules were found to be significantly correlated with treatment response. Of these 176 DEGs, 79 DEGs (M_DEGs) were the members of these 22 co-expression modules. Of the 287 GO functional processes and pathways that were enriched for these 79 M_DEGs, we identified 30 pathways whose overall gene expression activities were significantly correlated with treatment response. Of the original 176 DEGs, 19 (GO_DEGs) were found to be the members of these 30 pathways, whose expression profiles showed clear discrimination before and after treatment. As expected, of the biological processes and functionalities implicated by these 30 treatment response-related pathways, the inflammation and immune response was the top pathway in response to etanercept treatment, and some known TNF- α related pathways, such as molting cycle process, hair cycle process, skin epidermis development, regulation of hair follicle development, were implicated. Furthermore, additional novel pathways were also suggested, such as heparan sulfate proteoglycan metabolic process, vascular endothelial growth factor production, whose transcriptional regulation may mediate the response to etanercept treatment.
Conclusions
Through global gene expression analysis in PBMC of psoriasis patient and subsequent co-expression module based pathway analyses, we have identified a group of functionally coherent and differentially expressed genes (DEGs) and related pathways, which has not only provided new biological insight about the molecular mechanism of anti-TNF- α treatment, but also identified several genes whose expression profiles can be used as potential biomarkers for anti-TNF- α treatment response in psoriasis.
Brief:anti-TNF-α对银屑病的治疗分子机制研究,差异基因,WGCNA及GO annonation

Background
Identification of Hürthle cell cancers by non-operative fine-needle aspiration biopsy (FNAB) of thyroid nodules is challenging. Resultingly, non-cancerous Hürthle lesions were conventionally distinguished from Hürthle cell cancers by histopathological examination of tissue following surgical resection. Reliance on histopathological evaluation requires patients to undergo surgery to obtain a diagnosis despite most being non-cancerous. It is highly desirable to avoid surgery and to provide accurate classification of benignity versus malignancy from FNAB preoperatively. In our first-generation algorithm, Gene Expression Classifier (GEC), we achieved this goal by using machine learning (ML) on gene expression features. The classifier is sensitive, but not specific due in part to the presence of non-neoplastic benign Hürthle cells in many FNAB.
Results
We sought to overcome this low-specificity limitation by expanding the feature set for ML using next-generation whole transcriptome RNA sequencing and called the improved algorithm the Genomic Sequencing Classifier (GSC). The Hürthle identification leverages mitochondrial expression and we developed novel feature extraction mechanisms to measure chromosomal and genomic level loss-of-heterozygosity (LOH) for the algorithm. Additionally, we developed a multi-layered system of cascading classifiers to sequentially triage Hürthle cell-containing FNAB, including: 1. presence of Hürthle cells, 2. presence of neoplastic Hürthle cells, and 3. presence of benign Hürthle cells. The final Hürthle cell Index utilizes 1048 nuclear and mitochondrial genes; and Hürthle cell Neoplasm Index leverages LOH features as well as 2041 genes. Both indices are Support Vector Machine (SVM) based. The third classifier, the GSC Benign/Suspicious classifier, utilizes 1115 core genes and is an ensemble classifier incorporating 12 individual models.
Conclusions
The accurate algorithmic depiction of this complex biological system among Hürthle subtypes results in a dramatic improvement of classification performance; specificity among Hürthle cell neoplasms increases from 11.8% with the GEC to 58.8% with the GSC, while maintaining the same sensitivity of 89%.
Brief:通过机器学习对病理情况下特定细胞的识别

Background
Biological experiments have confirmed the association between miRNAs and various diseases. However, such experiments are costly and time consuming. Computational methods help select potential disease-related miRNAs to improve the efficiency of biological experiments.
Results
FCMDAP achieves an average AUC of 0.9165 based on leave-one-out cross validation. Results confirm the 100, 98 and 96% of the top 50 predicted miRNAs reported in case studies on colorectal, lung, and pancreatic neoplasms. FCMDAP also exhibits satisfactory performance in predicting diseases without any related miRNAs and miRNAs without any related diseases.
Conclusions
In this study, we present a computational method FCMDAP to improve the prediction accuracy of disease related miRNAs. FCMDAP could be an effective tool for further biological experiments.
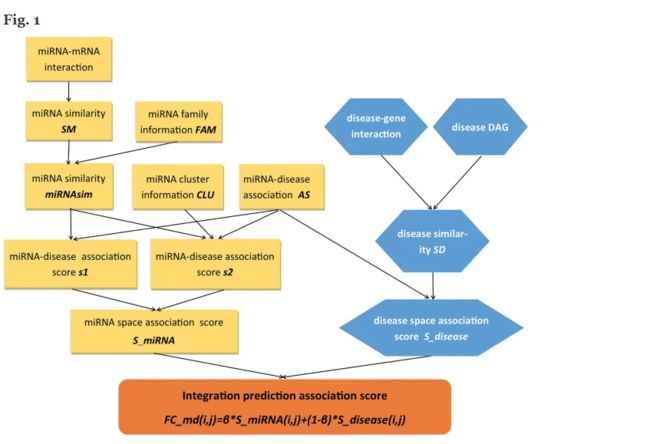
Brief:基于mRNA及miRNA互作及miRNA基因家族数据利用互信息来衡量miRNA相关性;同时根据疾病基因互作获得疾病之间功能相似性,同时用这两种度量方式来预测潜在的疾病相关的miRNA

Background
Single-cell RNA sequencing (scRNA-Seq) is an emerging technology that has revolutionized the research of the tumor heterogeneity. However, the highly sparse data matrices generated by the technology have posed an obstacle to the analysis of differential gene regulatory networks.
Results
Addressing the challenges, this study presents, as far as we know, the first bioinformatics tool for scRNA-Seq-based differential network analysis (scdNet). The tool features a sample size adjustment of gene-gene correlation, comparison of inter-state correlations, and construction of differential networks. A simulation analysis demonstrated the power of scdNet in the analyses of sparse scRNA-Seq data matrices, with low requirement on the sample size, high computation efficiency, and tolerance of sequencing noises. Applying the tool to analyze two datasets of single circulating tumor cells (CTCs) of prostate cancer and early mouse embryos, our data demonstrated that differential gene regulation plays crucial roles in anti-androgen resistance and early embryonic development.
Conclusions
Overall, the tool is widely applicable to datasets generated by the emerging technology to bring biological insights into tumor heterogeneity and other studies. MATLAB implementation of scdNet is available at https://github.com/ChenLabGCCRI/scdNet.
Brief:单细胞的稀疏特性给基因调控网络分析带来困难,因此提出该算法,提出一种细胞状态前的差异基因调控网络的方法,不同的state独立归一化并且消除样本大小的影响后独立进行相关性分析,两种状态的相关性网络构建好后进行费舍尔精确检验,然后将有差异的的基因对保留产生细胞状态间的差异调控网络

Background
Adenocarcinoma in situ (AIS) is a pre-invasive lesion in the lung and a subtype of lung adenocarcinoma. The patients with AIS can be cured by resecting the lesion completely. In contrast, the patients with invasive lung adenocarcinoma have very poor 5-year survival rate. AIS can develop into invasive lung adenocarcinoma. The investigation and comparison of AIS and invasive lung adenocarcinoma at the genomic level can deepen our understanding of the mechanisms underlying lung cancer development.
Results
In this study, we identified 61 lung adenocarcinoma (LUAD) invasive-specific differentially expressed genes, including nine long non-coding RNAs (lncRNAs) based on RNA sequencing techniques (RNA-seq) data from normal, AIS, and invasive tissue samples. These genes displayed concordant differential expression (DE) patterns in the independent stage III LUAD tissues obtained from The Cancer Genome Atlas (TCGA) RNA-seq dataset. For individual invasive-specific genes, we constructed subnetworks using the Genetic Algorithm (GA) based on protein-protein interactions, protein-DNA interactions and lncRNA regulations. A total of 19 core subnetworks that consisted of invasive-specific genes and at least one putative lung cancer driver gene were identified by our study. Functional analysis of the core subnetworks revealed their enrichment in known pathways and biological progresses responsible for tumor growth and invasion, including the VEGF signaling pathway and the negative regulation of cell growth.
Conclusions
Our comparison analysis of invasive cases, normal and AIS uncovered critical genes that involved in the LUAD invasion progression. Furthermore, the GA-based network method revealed gene clusters that may function in the pathways contributing to tumor invasion. The interactions between differentially expressed genes and putative driver genes identified through the network analysis can offer new targets for preventing the cancer invasion and potentially increase the survival rate for cancer patients.
Brief:原位腺癌与侵袭性腺癌的转录组研究,是否可做侵袭性肺癌的预测?

Background
Single-cell RNA sequencing is a powerful tool for characterizing cellular heterogeneity in gene expression. However, high variability and a large number of zero counts present challenges for analysis and interpretation. There is substantial controversy over the origins and proper treatment of zeros and no consensus on whether zero-inflated count distributions are necessary or even useful. While some studies assume the existence of zero inflation due to technical artifacts and attempt to impute the missing information, other recent studies argue that there is no zero inflation in scRNA-seq data.
Results
We apply a Bayesian model selection approach to unambiguously demonstrate zero inflation in multiple biologically realistic scRNA-seq datasets. We show that the primary causes of zero inflation are not technical but rather biological in nature. We also demonstrate that parameter estimates from the zero-inflated negative binomial distribution are an unreliable indicator of zero inflation.
Conclusions
Despite the existence of zero inflation in scRNA-seq counts, we recommend the generalized linear model with negative binomial count distribution, not zero-inflated, as a suitable reference model for scRNA-seq analysis.
Brief:单细胞数据的零膨胀问题,认为零膨胀主要由生物学差异解释;认为不应通过imputation的方法补全数据

Abstract
We present Model-based AnalysEs of Transcriptome and RegulOme (MAESTRO), a comprehensive open-source computational workflow (http://github.com/liulab-dfci/MAESTRO) for the integrative analyses of single-cell RNA-seq (scRNA-seq) and ATAC-seq (scATAC-seq) data from multiple platforms. MAESTRO provides functions for pre-processing, alignment, quality control, expression and chromatin accessibility quantification, clustering, differential analysis, and annotation. By modeling gene regulatory potential from chromatin accessibilities at the single-cell level, MAESTRO outperforms the existing methods for integrating the cell clusters between scRNA-seq and scATAC-seq. Furthermore, MAESTRO supports automatic cell-type annotation using predefined cell type marker genes and identifies driver regulators from differential scRNA-seq genes and scATAC-seq peaks.
Brief:scRNA-seq&scATAC-seq整合分析工具

Background
The rapid development of single-cell RNA-sequencing (scRNA-seq) technologies has led to the emergence of many methods for removing systematic technical noises, including imputation methods, which aim to address the increased sparsity observed in single-cell data. Although many imputation methods have been developed, there is no consensus on how methods compare to each other.
Results
Here, we perform a systematic evaluation of 18 scRNA-seq imputation methods to assess their accuracy and usability. We benchmark these methods in terms of the similarity between imputed cell profiles and bulk samples and whether these methods recover relevant biological signals or introduce spurious noise in downstream differential expression, unsupervised clustering, and pseudotemporal trajectory analyses, as well as their computational run time, memory usage, and scalability. Methods are evaluated using data from both cell lines and tissues and from both plate- and droplet-based single-cell platforms.
Conclusions
We found that the majority of scRNA-seq imputation methods outperformed no imputation in recovering gene expression observed in bulk RNA-seq. However, the majority of the methods did not improve performance in downstream analyses compared to no imputation, in particular for clustering and trajectory analysis, and thus should be used with caution. In addition, we found substantial variability in the performance of the methods within each evaluation aspect. Overall, MAGIC, kNN-smoothing, and SAVER were found to outperform the other methods most consistently.
Brief:对目前现有的imputation方法进行了综合评估,结果提示经过推算后的数据恢复了部分的基因表达情况,但大部分方法并不能增效下游分析,各方法中MAGIC,kNN-smoothing及SAVER表现相对较好,imputation的方法对于差异分析可能更有效,而不是探索细胞间关系

Brief:单细胞数据跨实验及样本数据整合分析工具

Brief:singlecell-RNA-seq标准化pipeline

Abstract
We introduce Proteome-Wide Association Study (PWAS), a new method for detecting gene-phenotype associations mediated by protein function alterations. PWAS aggregates the signal of all variants jointly affecting a protein-coding gene and assesses their overall impact on the protein’s function using machine learning and probabilistic models. Subsequently, it tests whether the gene exhibits functional variability between individuals that correlates with the phenotype of interest. PWAS can capture complex modes of heritability, including recessive inheritance. A comparison with GWAS and other existing methods proves its capacity to recover causal protein-coding genes and highlight new associations. PWAS is available as a command-line tool.
Brief:全蛋白质组关联研究

Abstract
Dropouts distort gene expression and misclassify cell types in single-cell transcriptome. Although imputation may improve gene expression and downstream analysis to some degree, it also inevitably introduces false signals. We develop DISC, a novel deep learning network with semi-supervised learning to infer gene structure and expression obscured by dropouts. Compared with seven state-of-the-art imputation approaches on ten real-world datasets, we show that DISC consistently outperforms the other approaches. Its applicability, scalability, and reliability make DISC a promising approach to recover gene expression, enhance gene and cell structures, and improve cell type identification for sparse scRNA-seq data.
Brief:基于深度学习及本监督学习开发的inputation方法,测试数据集中促进了差异基因的发现及细胞类型鉴定

Abstract
High-throughput single-cell RNA-seq (scRNA-seq) is a powerful tool for studying gene expression in single cells. Most current scRNA-seq bioinformatics tools focus on analysing overall expression levels, largely ignoring alternative mRNA isoform expression. We present a computational pipeline, Sierra, that readily detects differential transcript usage from data generated by commonly used polyA-captured scRNA-seq technology. We validate Sierra by comparing cardiac scRNA-seq cell types to bulk RNA-seq of matched populations, finding significant overlap in differential transcripts. Sierra detects differential transcript usage across human peripheral blood mononuclear cells and the Tabula Muris, and 3 ′UTR shortening in cardiac fibroblasts. Sierra is available at https://github.com/VCCRI/Sierra.
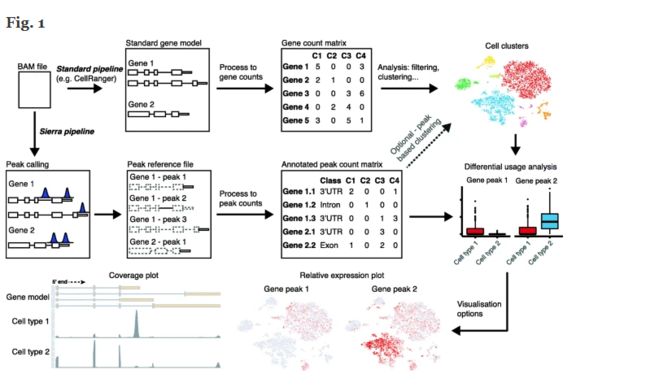
Brief:单细胞转录本亚型鉴定及定量化分析software

Summary
To what extent stromal cells in the tumor microenvironment (TME) are transformed by colorectal cancer (CRC) cells is unexplored. To dissect alterations in these non-malignant cells, we performed single-cell multiomics sequencing of 21 patients with microsatellite-stable CRCs and 6 cancer-free, elderly individuals. Surprisingly, somatic copy number alterations (SCNAs) are prevalent in immune cells, fibroblasts, and endothelial cells in both the TME and the normal tissues of each individual. Moreover, the proportions of fibroblasts with SCNAs in tumors (11.1%–47.7%) are much higher than those in adjacent normal tissues (1.1%–10.6%), with gain of chromosome 7 strongly enriched in the TME, clearly indicating clonal expansion. Furthermore, five genes (BGN, RCN3, TAGLN, MYL9, and TPM2) are identified as fibroblast-specific biomarkers of poorer prognosis of CRC. Our study provides evidence and functional relevance of pervasive genomic alterations in the stromal cells of TME in CRC.
Brief:通过单细胞基因组及转录组结合测序的方法的结直肠癌患者的单细胞多组学研究,发现体细胞拷贝数改变在每个个体的正常细胞都普遍存在,但携带体细胞拷贝数改变的成纤维细胞在肿瘤中富集,提示其在癌症发生发展的作用