今天,咱们来看一篇7月末发表在Aging-US(IF=4.831)杂志上的一篇文章,作者基于TCGA中的NSCLC(非小细胞肺癌)数据,识别出了4个预测复发的CpG甲基化markers,并验证了这一特征与TMB(肿瘤突变负荷)和DDR(DNA损伤应答)之间的显著关联,接下来详细了解一下吧!
Identifying CpG methylation signature as a
promising biomarker for
recurrence and immunotherapy in non–small-cell lung carcinoma
识别NSCLC复发和免疫治疗的CpG甲基化标志物
数据和方法
NSCLC患者数据
研究使用到的数据有包含901 个TCGA样本的level3的DNAm数据;还有,GEO中的三套甲基化数据集GSE66836、GSE39279和GSE119144及其相应的临床数据;TCGA中包含1007个NSCLC组织和109个正常组织的基因表达谱数据及相关的临床数据;TCGA中NSCLC的体细胞突变数据。大部分数据下载自https://tcga.xenahubs.net。
数据集的分析以及构建预测模型
首先,作者用R中的limma包对TCGA level3 DNA甲基化数据筛选了差异甲基化位点(DMPs),BH矫正后的显著性P值<0.05,就认为相应< span="">DMP包含CpG。此外,作者又对GSE6683的450K甲基化数据做了相同分析,提取出了两个数据集重合的DMPs。log2FC<0的dmps为低甲基化,>0则是高甲基化。对于450K数据,作者用R包lluminaHumanMethylation450kanno.ilmn12.hg19做了注释。
然后,用R包edgeR分析了有甲基化数据的821例TCGA NSCLC样本和28例匹配的正常肺组织样本,FDR << span="">0.05且|log2FC| > 1的为差异表达基因(DEGs)。其中,低甲基化CpGs表现出较高的表达,高甲基化CpGs的表达水平则较低。接下来,作者整合了mRNA表达谱和表观遗传谱,用Spearman相关评估了位于转录起始位点2kb内的DEGs与DMPs之间的关联,筛选阈值为Bonferroni校正的P<0.05和r<0< span="">。
为了识别可预测NSCLC复发的甲基化标志物,TCGA肺癌群体被用来做训练集,664例NSCLC样本具有无复发生存(RFS)信息。首先,通过随机森林和LASSO回归模型识别复发相关的DNAm markers,并用LASSO-Cox对挑选出的CpGs做进一步的筛选;同时,利用单因素cox回归筛选与NSCLC患者RFS相关的CpG markers。然后,保留两个结果中相同的CpGs,来构建多变量cox回归模型。最后,三个GEO数据集用来做进一步的验证,其中,GSE119144包含了60例接受抗PD-1/PD-L1 ICBS治疗的NSCLC患者,59个样本有完整的随访信息。CpG marker的筛选流程如图1所示。

图1. 筛选CpG marker
基因集富集分析
为了探讨基于DNAm风险评分预测的NSCLC复发风险状态,和基因特征间的关联,作者用R包”clusterprofiler”对TCGA mRNA表达数据做了GSEA分析,根据风险评分中指患者被分成高、低风险组。此外,”GSVA”包用来识别DNAm特征相关的通路,对186个KEGG通路特征进行了基因集变异分析(GSVA),P < 0.05的基因特征认为是显著差异富集的。以上两个参考基因集下载自MsigDB数据库。
体细胞突变分析
研究分析了1048 例TCGA NSCLC样本中的高置信的体细胞变异,包括单核苷酸变异和短的插入/缺失,然后,用MutsigCV分析显著突变的基因(SMGs),保留FDR<< span="">0.05的;接下来,用R包Maftool实现了TCGA NSCLC全外显子测序(WES)数据的MAF文件的可视化和概要部分;为评估肿瘤突变负荷(TMB),又计算了每个肿瘤样本编码区的非同义体细胞突变。对于7种免疫细胞亚型(B细胞、NK细胞、CD4+和CD8+ T细胞、单核细胞、性粒细胞和嗜酸性粒细胞),作者对TCGA DNAm数据和GSE66836集使用R包EpiDISH”,来推断NSCLC患者中包含的单个细胞类型分数。随后,分析了DNAm特征与估计的细胞类型富集之间的关系。
统计分析
所有统计分析都是通过R完成的。对筛选出的DMPs的DNA甲基化水平以及附近DEGs的mRNA表达水平的无监督层次聚类,使用的是R包ComplexHeatmap。R包“randomForest”和“glmnet”分别用于随机森林和LASSO模型,10倍交叉验证用于模型的优化。KM曲线和log-rank检验用的是survminer包,在DNAm特征和临床参数结合的多变量Cox比例风险回归模型中,该包也被用来做生存分析。pROC包用于评估CpG markers在训练集和验证集中的效能。Wilcoxon用来比较组间差异。采用Spearman相关分析评估DNAm特征与预估的细胞型组分、DNA损伤反应(DDR)基因和蛋白质的关系。两个风险组之间的DDR基因突变频率用卡方检验进行比较。对于所有检验,双侧P < 0.05为显著性阈值。
结果
NSCLC的DNA甲基化和基因表达谱
针对TCGA和GSE66836数据集,作者分析了NCSLC和正常肺组织样本之间的DNAm差异,识别出了11641个重合的差异CpGs,其中,在另外两个验证集中也显著差异的有9367个。然后,作者对TCGA的RNA-seq数据做了差异表达分析,识别出了1717个DEGs,根据这些基因甲基化程度以及与上述DMPs的位置关系,进一步筛选了CpGs。在对DNAm和mRNA数据做了Spearman检验后,最终保留了102个CpGs,对应着87个DEGs。接下来,对这87个DEGs的无监督层次聚类将TCGA样本分成了肿瘤和正常两组,肿瘤样本中有53个基因上调以及34个基因下调(图2.A)。对102个DMPs的聚类也得到了相似的结果,在NSCLC样本中,有57个低甲基化,45个高甲基化(图2.B)。
图2. 无监督层次聚类
识别可预测NSCLC的CpGs
首先,作者分别用LASSO-Logistic回归和随机森林两种方法对前面提到的102个CpGs进行建模,各自识别出14和21 个CpGs markers(图3.A-B),交集有11个,并集24个。这24个CpGs通过LASSO-Cox建模,筛选出了9个稳健的预后CpG标志物(图3.C-D)。同时,作者用训练集的无复发生存数据进行单变量Cox回归分析,筛选出8个CPGS,与用LASSO-Cox的筛选结果相同的显著CpG标志物有4个(图3.E),这两种方法共识别出13个预测标志物。基于以上4个CpGs,作者构建了一个风险评分模型,用于预测NSCLC的复发,并根据风险评分中值将NSCLC患者分为高、低风险组。
研究进一步分析了该风险评分模型预测NSCLC预后的潜力,结合了风险评分的生存分析表明,高、低危险组的NSCLC患者RFS概率存在显著差异,而4-DNAm-marker在预测TCGA NSCLC患者的总生存期时也具有良好的潜能(图3F)。
此外,多变量生存分析结果表明,该风险评分可作为独立的NSCLC预后因子,且与RFS和OS均显著相关。

图3. 风险评分模型
风险评分相关的临床及分子特征
研究发现,复发患者的风险评分显著更高,不同分期的TCGANSCLC患者,复发风险也显著不同,两个GEO数据集也得到了相似的结果(图4.A)。对TCGA风险亚组的GSEA分析发现,高危组显著富集的基因特征主要与E2F靶点、G2M检查点和MYC靶点V1有关(图4.B)。对TCGA中LUAD患者的RPPA分析表明,较高的风险评分与FOXM1和CYCLINB1蛋白的高表达显著相关。如图4.B所示,该4-DNAm特征显著富集到的通路主要与细胞周期、增殖和免疫相关,so,作者在TME(肿瘤微环境)的背景下评估了基于DNAm的风险评分,然后,发现高风险评分与成纤维细胞的增加和免疫细胞分数的降低有关(图4.C)。
接下来,基于TCGA NSCLC的突变谱,作者用MutsigCV识别出了几个与风险评分相关的SMGs,如图4.D所示。基因KRAS, KEAP1, STK11和KRAS/KEP1A的突变均与风险评分明显相关(图4.E),图4.F列出了几个表达与风险评分相关的基因。
图4. 风险评分相关的因素
甲基化特征与TMB和DDR基因相关
基于与TCGA甲基化数据匹配的体细胞突变数据中的非同义编码突变,作者分析了DNAm特征与TMB的关联关系,发现高风险组的TMB也更高(图5.A)。用来构建风险模型的这4个DMPs的甲基化状态以及附近基因的表达水平,同样与TMB相关。基因集富集结果表明了细胞增殖过程的激活与高风险状态间的关联,同时DNAm特征可能与细胞周期、DNA复制和DDR通路基因的改变有关,对DDR通路的进一步分析,发现高风险组患者的TP53突变频率更高(图5.B)。所有分析表明,NSCLC中DNAm模式的改变可能会通过介导细胞周期调节和DDR基因的变异,来影响TMB,从而导致更多的新抗原形成和肿瘤抗原性的变化。

图5. 风险评分相关的基因
高风险评分患者免疫治疗效果更好
研究利用GSE119144数据集,分析了基于DNAm的风险评分与ICBs(免疫抑制阻断剂)反应间的关系,发现高风险组免疫治疗的RFS优于低风险组;而且,相较于低风险组,较大比例的NSCLC患者具有更持久的临床效益(图6.A-B)。
值得注意的是,DNAm特征和TMB的结合显著提高了预测免疫治疗临床反应的效能(图6.C),从图6.D中的Kaplan-Meier曲线也可看出,由这两个变量组合分组的NSCLC患者具有显著不同的临床结果。
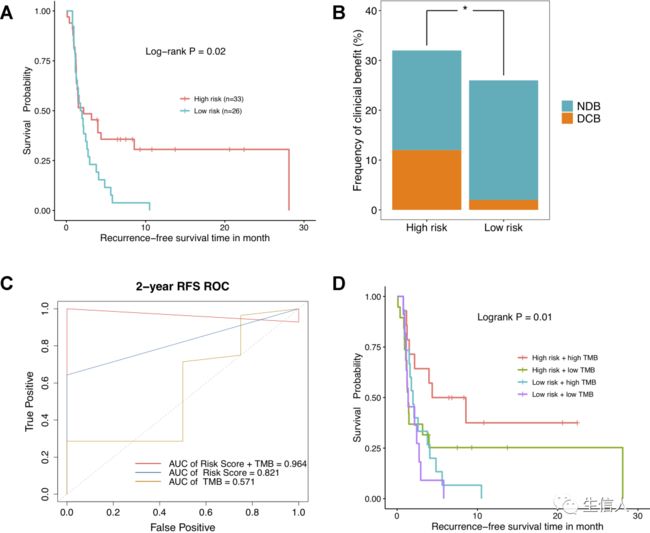
图6. DNA甲基化与免疫反应
总结一下,有分析意向(http://gaptechsxr.mikecrm.com/1vdMmqy)生信人WX公众号作者筛选出了一个DNAm特征(包含cg00253681、cg00111503、cg02715629和cg03282991),在预测NSCLC复发和ICBs治疗的临床效益方面都具有良好的效能,揭示了表观遗传调控在疾病复发预测、治疗策略选择和免疫治疗反应评估中的意义。