目录
- 背景知识
- 1.1. 根据分析对象的分类
- 1.2. 三个基本数据处理问题
- 1.3. 一般分析流程
- 1.4. 比较宏基因组学的应用
- 1.5. 目前存在的技术问题
- 实验设计
- 2.1. 几点指导意见
- 2.2. 测序平台的选择
- Metagenome assembly
- 宏基因组binning
- 4.1. binning原理
- 4.2. binning具体操作
- 4.3. 目前binning工具存在的问题
- Taxonomic profiling
- 5.1. Assembly-free strategy
- 5.2. 比较 Assembly-based profiling 与 Read-based profiling
- Metabolic function profiling

1. 背景知识
1.1. 根据分析对象的分类
根据分析对象和实验目的,宏基因组的研究基本上可以分为
- 扩增子测序:
核糖体rDNA(细菌和古细菌 16S rDNA 或真菌 18S、28S rDNA 和 ITS (Internal Transcribed Spacer,真菌 rDNA 基因非转录区的一部分))的分类和鉴定
获得环境中各个细菌种类的相对丰度和多样性水平,从而了解环境中微生物群落的组成和结构
single marker genes(一般为功能基因,比如固氮还原酶nifH基因和氨基氧化酶amoA基因等)的多样性和分类分析
揭示各个功能菌群的构成和多样性
- 宏基因组全测序 Shotgun metagenomics:全部宏基因组DNA的整体测序和分析
- profile taxonomic composition
- functional potential of microbial communities
- to recover whole genome sequences
1.2. 三个基本数据处理问题

1.3. 一般分析流程

1. Pre-processing
- minimize fundamental sequence biases or artifacts such as removal of sequencing adaptors
- quality trimming
- removal of sequencing duplicates
- filter foreign or non-target DNA sequences
- samples are subsampled to normalize read numbers if the diversity of taxa or functions is compared
2. Sequence analysis
包括两种分析策略:read-based (mapping) 和 assembly-based

简单来说,assembly-based approach 受到覆盖度的制约,因为组装时低覆盖度的区域是不会进行组装的,而是被丢弃,这样低丰度的细菌的信息就被丢弃了,反映在reads利用率上,就是往往reads利用率极低,往往低于50%
而 read-based (mapping) approach 则受到reference databases的制约,因为细菌的遗传多样性很高,即便是同一个菌种,它的不同菌株,其基因组的组成也是有相对比较大的差异的,那么在mapping的时候就会出现mapping不上的问题,使得mapping效率不够高;而且只能分析reference databases中有的物种,对于reference databases未收录的新物种,是无法进行分析的。
不过可用的微生物参考基因组正在迅速地增加,包括那些原先难以培养的细菌由于培养方法的改进,使得对其进行测序成为可能,再加上单细胞测序的途径和 metagenomic assembly的途径得到的基因组序列。现在一些类型的环境样品(如人肠道)的参考基因组的多样性已经可以满足 assembly-free taxonomic profiling 的要求。
1.4. 比较宏基因组学的应用
- 了解微生物群体结构随着时间和地理环境的变化,以及通过比较不同个体、不同时间点、不同药物作用时微生物的变化与疾病的相关性
1.5. 目前存在的技术问题
- 分析难度大,分析成本高
随着测序成本的下降和测序深度的增加,其分析难度将会越来越大,制约效应也将会越来越明显
预计的单位测序成本将会以指数关系下降,但其中计算成本下降的幅度会远慢于测序成
- 宏基因组全测序的分析主要受限于计算技术的发展
在数据存储和数据处理的层面上,rDNA和扩增序列的分析难度较小,基本可以在个人电脑或者小型服务器上完成,但宏基因组全测序的分析却主要受限于计算技术的发展
数据存储的容量
原始测序数据量本来就不小,而随后的分析又会产生10倍以上的数据量
序列的拼接
目前比较成熟的序列拼接算法都是基于一个或少数几个基因组的数据(如 Genovo,MetaVelvet,MAP等),而对 HiSeq 2000 所产生的宏基因组数据无能为力,其最主要的原因是所有的拼接算法都需要庞大的内存资源,而这一需求远大于市面上最大的单服务器所支持的内存数量(4 Tb 左右)
序列拼接、基因和基因功能预测的准确性同速度之间的矛盾
拼接和基因预测算法往往需要耗费大量的运算资源,而很多近似或高速算法往往以牺牲准确性作为代价。因此,如何在保证准确性的前提下提高速度是决定宏基因组分析质量的关键
- 数据挖掘的层面上存在难点
物种多样性(Taxonomy diversity)、功能多样性 (Functional diversity) 和遗传多样性(Genetic diversity)的估算
因为稀有物种的大量检出,经典的估算方法如 Chao 等都会产生严重的偏差
宏观生态理论在分子生态中的运用
现代生态学经过 20 世纪的发展已经积累了大量成熟的理论和模型,然而大部分的生态群落理论还是建立在宏观生态的基础之上,这些理论是否也适用于微观领域,现在还没有明确的结果支持
微生物物种间关联的不确定性
生物群落的结构不仅包括多样性和物种数量上的分布,而且应该包含物种间的相互作用关系,而这些关系在物质、能量、信息循环中起到了至关重要的作用。然而目前微生物物种间的相互作用往往法像宏观生态中予以观察和定性
2. 实验设计
即使在同一个环境中获取的不同样本,其微生物组成也会存在比较大的差异,这使得在样本集之间,寻找具有统计学显著性和生物学意义的差异变得很困难。因此如何做到,在即使其影响因素的作用程度很小的情况下,也能有效地检测出差异就显得十分重要。
- 评估实验与分析结果的可靠性
一种策略是,构造 pilot data,即将不同浓度的绝对定量 control (spike-in) 加入到样本中,来评估实验与分析方法的稳健性(robust);
另一种策略:two-tiered approach,即挑取少部分样本,既做 16s rDNA 测序,又做 shotgun metagenomics 测序,对比这两个层次的结果来评估实验结果的稳健性。
- 横向/纵向 研究策略的比较
两种研究策略:
- cross-sectional studies —— 横向研究,又叫横断研究或横向比较研究,就是在同一时间内对每个对象进行观察与测定,在相互比较的基础上对特定因素或各种因素间的关系进行分析与考察的研究方案
- longitudinal studies —— 纵向追踪研究,亦称作纵向研究或追踪研究,是在比较长的时间内对相同对象进行有系统的定期研究,或者从时间的发展过程中考察研究对象的研究方案
由于在研究宏基因组过程中,比如研究人类的微生物群,影响其微生物群的因素众多,包括宿主基因型,年龄,饮食习惯等等,当进行两个环境微生物群横向比较时,很难做到控制变量,使得在进行比较分析时混入了许多干扰因素;此时如果进行单一环境微生物群多时间点采样的纵向比较,就可以从很大程度上消除这种影响。
2.1. 几点指导意见
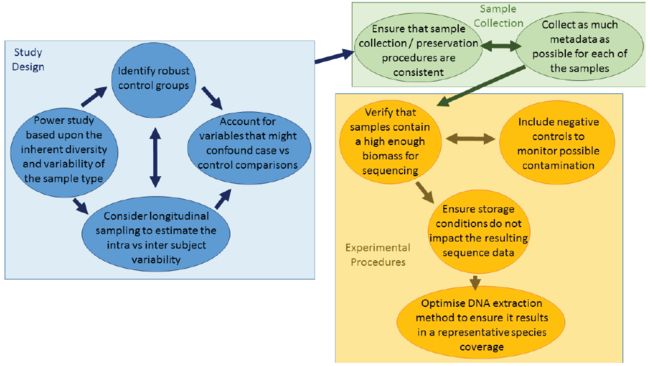
1. 样本量与测序深度
当实验目的是检出显著性差异时,样本量与测序深度的选择取决于(1)不同样本间微生物组组成的一致性,(2)样本固有的微生物多样性,(3)影响因素的效应量(effect size)
建议:参考前人在类似环境中的研究。若没有可参照的类似研究,选择marker gene做预实验
2. Confounding variables and control groups
在进行宏基因组研究时,往往很难找到与目标样本集对应的没有其他干扰因素的对照组
建议:目前最佳的解决策略是,尽可能地搜集各个样本群体的元数据 (metadata),然后在随后的比较分析中将它们考虑进去。比如临床样本,包括性别、年龄、是否使用抗生素/药物、取样位置、饮食习惯等等。比如环境样本,包括地理位置、季节、pH、温度等等。
元数据的搜集可以参照MIMARKS (Minimum information about a marker gene sequence) 和 MIxS (minimum information about any (x) gene sequence) 标准
3. Sample collection/preservation
样本的处理和保存过程的差异会带来系统偏差,比如when samples are provided from a number of locations by different research groups,或者在纵向研究中,不同取样时间点的样本的保存时间长短不一。有时这些处理步骤的效应量可能比你感兴趣的生物学变量还大。
建议:尽可能按照相同的标准来进行取样和保存
4. Biomass/Contamination
当前采用的基于测序的方法具有很高的灵敏度 (highly sensitive),即使非常微量的DNA也能被检测出来。而实验室中使用到的常规仪器和试剂并不是无菌的,这样就很可能在实验操作过程中,人为地引入污染。由于检测方法的高灵敏度,当原样本的微生物量很少时,污染带来的信号很可能会盖过真实的信号。
建议:在上机测序前,做好微生物量的定量 (qPCR)。当样品中的微生物数量少于105数量级时,其极有可能会受到背景污染的干扰。此时,可以参照以下的方法进行细胞/DNA的富集:

可以增设负对照实验 (Negative control),对其进行与实际样本相同的操作,使用相同的试剂,以此来找出污染的细菌类型,这样就可以在后续的生物信息学分析过程中将其过滤掉。
5. 选择合适的DNA提取方法
DNA提取的效果会直接对后续的实验和分析产生巨大的影响。DNA提取方法的选择依赖于样品中细胞类型的组成,然而即使是相同类型的样品其微生物组成也具有较大的差异(当人粪便中革兰氏阴性菌主导时,细胞很容易裂解,而当由相对顽强的革兰氏阳性菌主导时,则相反)。
因此不存在适用于所有样品的最佳的DNA提取方案。
若方案选择不当,则获得的DNA主要来自于那些易裂解的细菌
建议:
2.2. 测序平台的选择
Illumina测序仪通量大 (up to 1.5 Tb per run),且准确率高 (with a typical error rate of 0.1–1%),通过在不同样本的序列上添加两重barcode,可以一次测序多个samples。
然而,Illumina测序仪存在carryover (between runs) 和 carry-between (within runs)的问题。最新的测序仪由于使用了新的扩增方法 (ExAmp),导致较高比例的‘index hopping’。
虽然没有一个明确的指导意见,告诉你在哪个特定的环境样品中应该测多大的覆盖度,但是一个基本的原则就是通量要尽可能地大,这样低丰度的细菌也能被测到。Illumina HiSeq 2500/4000, NextSeq 和 NovaSeq 的测序通量都很大,都适用于 metagenomics 的研究。
HiSeq 2500 在 rapid-run 模式下能产生 2 × 250-nt 的 reads (up to 180 Gb per flow cell),在 high-output 模式下能产生 2 × 125-nt 的 reads (up to 1 Tb)
新一些的HiSeq 3000 和 4000,通量提高 (up to 1.5 Tb for the 4000),但是测序长度限制在 150nt
NextSeq 的通量与 HiSeq 2500 的 high-output 模式相同,而花费只有Hiseq的一半,但是长度限制为 150nt
最近才推出的 Novaseq 有望达到 3 Tb per flow cell
MiSeq 受制于它的通量 (up to 15 Gb in 2 × 300 mode),但仍然是目前 single-marker-gene microbiome studies 的金标准
3. Metagenome assembly
Metagenome de novo assembly 采用的策略与 whole-genome assembly 相同,均为 de Bruijn 图方法
用 de Bruijn 图方法进行宏基因组的从头组装时,面临着以下的挑战:
- 测序覆盖度不均匀
当进行单一基因组的组装时,其有一个前提假设:整个基因组的测序覆盖度是相对均匀的,这样就可以利用覆盖度信息来识别重复序列和鉴定测序错误和等位变异。
而metagenome中,各个组成基因组的覆盖度取决于它们的物种丰度,低丰度物种的基因组就会由于总体测序深度不够而使得最终组装出来的基因组是支离破碎的。使用更短的 k-mer 有助于低丰度基因组的组装,但是这会使得图中重复 k-mer 的频率大大增加,降低了组装的准确性。
这需要组装工具在考量低丰度物种与获得高丰度物种更长更准确的contig之间进行权衡,即选择合适的 k-mer :
If k is too large, there will be a lot of gap problems in the graph.
If k is too small, there will a lot of branch problems.
Meta-IDBA:使用多重 k-mer
IDBA-UD:基于Meta-IDBA的升级,对测序深度不均匀数据的组装过程进行了优化
- 同种细菌不同菌株的干扰
同种细菌的不同菌株,它们的基因组组成很相近,常常就是一个碱基的变异或者整个基因/操纵子的丢失,当进行 de Bruijn 图组装时,就会在这些差异的位置出现分叉,组装工具在遇到这些分叉时,常常会停在这些位置,从而导致一个个不连续组装片段的产生。
Meta-IDBA:将图依据其拓扑结构拆分成各个元件,每个元件代表各个亚种的共有区域
Meta-IDBA handles this problem grouping similar regions of similar subspecies by partitioning the graph into
components based on the topological structure of the graph. Each component represents a similar region between
subspecies from the same species or even from different species. After the components areseparated, all contigs
in it are aligned to produced a consensus and also the multiple alignment.
解决计算能力与内存不足的策略:
使用分布式 assemblers,例如 ABySS、Ray
将metagenome的组装图分割成相互连接的部分,然后在各个部分内部分别进行相对独立地组装,即分而治之的策略。
4. 宏基因组binning
Metagenome 组装完成后,我们得到的是成千上万的 contigs,我们需要知道哪些 contigs 来自哪一个基因组,或者都有哪些微生物的基因组。所以需要将 contigs 按照物种水平进行分组归类,称为 "bining"
Supervised binning methods: use databases of already sequenced genomes to label contigs into taxonomic classes
Unsupervised (clustering) methods: look for natural groups in the data
Both supervised and unsupervised methods have two main elements: a metric to define the similarity between a given contig and
a bin, and an algorithm to convert those similarities into assignments
一个很容易想到的策略就是,将组装得到的片段与已知物种的参考基因组进行比对,根据同源性进行归类。然而目前大多数的微生物的基因组还没有测序出来,因此限制了这种方法的可行性。
目前主流的 bining 策略利用的是 contigs 的序列组成特点。
4.1. binning原理

- 根据核酸组成信息来进行binning:k-mer frequencies
依据:来自同一菌株的序列,其核酸组成是相似的
例如根据核酸使用频率(oligonucleotide frequency variations),通常是四核苷酸频率(tetranucleotide frequency),GC含量和必需的单拷贝基因等
优势:即便只有一个样品的宏基因组数据也可以进行binning,这在原理上是可操作的
不足:由于很多微生物种内各基因型之间的基因组相似性很高,想利用1个样品的宏基因组数据通过核酸组成信息进行binning,效果往往并不理想或难度很大。利用核酸组成信息进行binning,基本上只适合那些群落中物种基因型有明显核酸组成差异的,例如低GC含量和一致的寡核苷酸使用频率
- 根据丰度信息来进行binning
依据:来自同一个菌株的基因在不同的样品中 ( 不同时间或不同病理程度 ) 的丰度分布模式是相似的【PMID: 24997787】。
原因:比如,某一细菌中有两个基因,A和B,它们在该细菌基因组中的拷贝数比例为 A:B = 2:1,则不管在哪个样品中这种细菌的数量有多少,这两个基因的丰度比例总是为 2:1
优势:这种方法更有普适性,一般效果也比较好,能达到菌株的水平
不足:必须要大样本量,一般至少要50个样本以上,至少要有2个组能呈现丰度变化 ( 即不同的处理,不同的时间,疾病和健康,或者不同的采样地点等 ) ,每个组内的生物学重复也要尽量的多
对于像质粒这样的可移动遗传单元 (mobile genetic elements (MGEs)),由于其复制独立于细菌染色体,则同一种细菌的不同个体,该质粒的拷贝数可能存在差异,使得无法用丰度信息进行有效地bining
- 同时依据核酸组成和丰度变化信息
将核酸组成信息和丰度差异信息创建一个综合的距离矩阵,既能保证binning效果,也能相对节约计算资源,现在比较主流的binning软件多是同时依据核酸组成和丰度变化信息
- 根据基因组甲基化模式
依据:不同的细菌,其基因组甲基化模式不同,平均一种细菌有3种特意的甲基化 motif。MGEs (mobile genetic elements) 中含有 MTase 基因,其基因水平转移是细菌甲基化组多样性的驱动因素。虽然 MGEs 在不同个体的拷贝数不同,但是都存在,因此具有相同 MGEs 的细菌个体,其总遗传物质(包括染色体和 MGEs )都会受到相同的MTase的作用而得到相同的甲基化模式。
4.2. binning具体操作
Q1:从哪些序列下手进行binning呢?
从原始的clean reads,还是从组装成的contig,还是从预测到的gene,都可以。根据基于聚类的序列类型的不同,暂且分为reads binning, contig binning和 genes binning
比较这三种binning的优劣:
contig binning
由于核酸组成和物种丰度变化模式在越长的序列中越显著和稳定,基于contig binning效果可能更好
reads binning
基于reads binning的优势是可以聚类出宏基因组中丰度非常低的物种
考虑到在宏基因组组装中reads利用率很低,单样品5Gb测序量情况下,环境样品组装reads利用率一般只有10%左右,肠道样品或极端环境样品组装reads利用率一般能达到30%,这样很多物种,尤其是低丰度的物种可能没有被组装出来,没有体现在gene 或者contig 中,因此基于reads binning 才有可能得到低丰度的物种
genes binning
应用非常广泛
原因可能是(1)基于genes丰度变化模式进行binning可操作性比较强,宏基因组分析中肯定都会计算gene丰度,一般不会计算contig丰度,gene丰度数据可以信手拈来;(2)基于genes binning有很多可参考的文献,过程也并不复杂,可复制性强;(3)对计算机资源消耗比较低
总体来说应用最广泛的就是基于genes binning 和 contig binning
Genes binning的一般流程
在宏基因组做完组装和基因预测之后,把所有样品中预测到的基因混合在一起,去冗余得到unique genes集合,对这个unique genes集合进行binning,主要是根据gene在各个样品中的丰度变化模式,计算gene之间的相关性,利用这种相关性进行聚类

该图中的聚类过程类似于K-means聚类:随机选择几个seed genes作为诱饵,计算其他基因丰度分布模式与seed genes的相关性,按照固定的相关性值PCC>0.9,将它们归属于不同seed genes所代表的类,然后在聚好的类内重新选择seed genes,进行迭代,最终聚类得到一个个基因集合,较大的集合(超过700个基因)称为 metagenomic species (MGS),较小的集合称为 co-abundance gene group (CAG)
基于 bining 结果进行单菌组装:
Sequence reads from individual samples that map to the MGS genes and their contigs are then extracted and used to assembly a draft genome sequence for an MGS
4.3. 目前binning工具存在的问题
- 还有很多可提升的空间
比如对核酸组成信息的利用,开发得就不够充分,四碱基使用频率因简单而被广泛使用和接受,但现在已有研究表明k-mer丰度信息也是很好的种系特征,同时越长的k-mer含有越多的信息,还有基因和参考基因组间的同源关系也是有价值的种系信号,但这些都还没有被自动化的binning软件整合
- 对于参数设置是很敏感的,且只有有限的可调整的参数
想要获得高质量的bins经常需要手动调整
5. Taxonomic profiling
Taxonomic profiling: identifies which microbial species are present in a metagenome and estimates their abundance
5.1. Assembly-free strategy
- reference-based approaches
优点:
mitigate assembly problems
speed up computation
enable profiling of low-abundance organisms that cannot be assembled de novo
当然它也有局限性:
previously uncharacterized microbes are difficult to profile
对于与人类密切相关的样品,比如人肠道,可以使用该策略,而且已经有相关的成功实践
- Marker-based approaches: by selecting representative or discriminative genes (markers)
By looking at co-abundant markers from preassembled environment-specific gene catalogs
即前人研究 (MetaHIT consortium) 已经得出特定环境下的微生物的组成,这些微生物中有某些 co-abundant markers(这些 marker genes 的丰度与其物种的丰度成正比),这样就可以基于对这些 markers 的定量得到对应的物种丰度
选择 markers 的不同策略:
mOTU: universally conserved but phylogenetically informative markers
MetaPhlAn: several thousand clade-specific markers
with high discriminatory power and was effective to quantitatively profile the microbiome from multiple body areas for the Human Microbiome Project (HMP) with a very low false positive discovery rate
当样本量巨大,都进行组装是明显不切实际的,此时采用 marker-based approaches 是一个不错的选择;而且,如果该环境来源的样本其组成微生物是研究比较充分时,marker-based approaches 能得到比较准确的物种定量结果。
5.2. 比较 Assembly-based profiling 与 Read-based profiling
| Assembly-based profiling | Read-based profiling |
|---|---|

|

|
6. Metabolic function profiling
Gene identification
homology-based annotation pipelines
translated sequence searches against functionally characterized protein families, such as KEGG or UniProt
Characterization of the functional potential of the microbiome
- 策略一:Aggregate single protein families into higher-level metabolic pathways and functional modules

Cleaned short DNA reads are aligned to the KEGG Orthology(or any other characterized sequence database) using accelerated translated BLAST.
Gene family abundances are calculated as weighted sums of the alignments from each read, normalized by gene length and alignment quality.
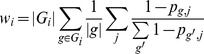
Assigning gene families to pathways and modules

Pathway reconstruction is performed using a maximum parsimony approach followed by taxonomic limitation (to remove false positive pathway identifications) and gap filling (to account for rare genes in abundant pathways).
The resulting output is a set of matrices of pathway coverages (presence/absence) and abundances
局限性: lack of annotations for accessory genes in most microbial species
因为在评估微生物群体的代谢潜能时,只对那些高度保守和 housekeeping 类型的功能进行了注释,这就解释了,为什么来自不同环境的不同样品,它们的功能特征常常是十分相似的,即使它们的物种组成有很大差异。
- 策略二:An in-depth characterization of specific functions of interest
例如,鉴定出微生物群落中的抗生素抗性基因,该方法高度依赖特定功能相关基因集注释的质量。
参考资料:
(1) 魏子艳, 金德才, 邓晔. 环境微生物宏基因组学研究中的生物信息学方法[J]. 微生物学通报, 2015, 42(5):890-901.
(2) Quince C, Walker A W, Simpson J T, et al. Shotgun metagenomics, from sampling to analysis[J]. Nature Biotechnology, 2017, 35(9):833.
(3) 句句干货!一文读懂宏基因组binning
(4) Nielsen H B, Almeida M, Juncker A S, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes[J]. Nature Biotechnology, 2014, 32(8):822-828.
(5) Sangwan N, Xia F, Gilbert J A. Recovering complete and draft population genomes from metagenome datasets[J]. Microbiome, 2016, 4(1):8.
(6) Abubucker, S. et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 8, e1002358(2012).
(7) Beaulaurier J, Zhu S, Deikus G, et al. Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation.[J]. Nature Biotechnology, 2017, 36(1).