USSC上的文件格式汇总
1.BED
某些翻译软件可能翻译为床文件,其实是叫Browser Extensible Data,直译就是浏览器拓展数据。
三个必须的列:The first three required BED fields are:
chrom - The name of the chromosome (e.g. chr3, chrY, chr2_random) or scaffold (e.g. scaffold10671).
chromStart - The starting position of the feature in the chromosome or scaffold. The first base in a chromosome is numbered 0.
chromEnd - The ending position of the feature in the chromosome or scaffold. The chromEnd base is not included in the display of the feature, however, the number in position format will be represented. For example, the first 100 bases of chromosome 1 are defined as chrom=1, chromStart=0, chromEnd=100, and span the bases numbered 0-99 in our software (not 0-100), but will represent the position notation chr1:1-100. Read more here.
关于染色体的起始位置和终止位置的坐标计数,The UCSC Genome Browser Coordinate Counting Systems有更多的解释。画的图很容易懂,用手指就可以领会。
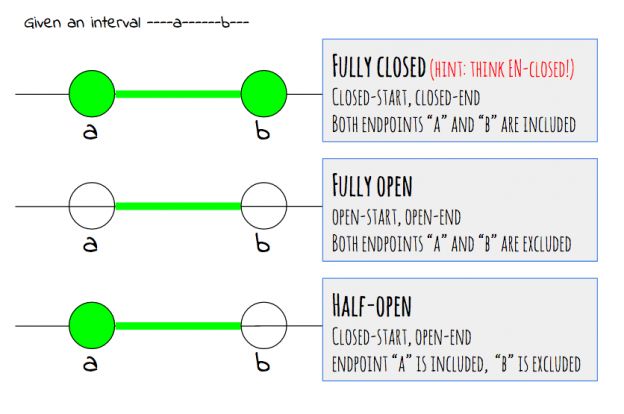


If you submit data to the browser in position format (chr#:##-##), the browser assumes this information is 1-based. If you submit data in any other format (BED (chr# ## ##) or otherwise), the browser will assume it is 0-based. Similarly, any data returned by the browser in position format is 1-based, while data returned in BED format is 0-based.至于为什么要有这两种文件格式,留坑,我也不知道。
The 9 additional optional BED fields are:
name - Defines the name of the BED line. This label is displayed to the left of the BED line in the Genome Browser window when the track is open to full display mode or directly to the left of the item in pack mode.
score - A score between 0 and 1000. If the track line useScore attribute is set to 1 for this annotation data set, the score value will determine the level of gray in which this feature is displayed (higher numbers = darker gray).
strand - Defines the strand. Either "." (=no strand) or "+" or "-".
thickStart - The starting position at which the feature is drawn thickly (for example, the start codon in gene displays). When there is no thick part, thickStart and thickEnd are usually set to the chromStart position.
thickEnd - The ending position at which the feature is drawn thickly (for example the stop codon in gene displays).
itemRgb - An RGB value of the form R,G,B (e.g. 255,0,0). If the track line itemRgb attribute is set to "On", this RBG value will determine the display color of the data contained in this BED line. NOTE: It is recommended that a simple color scheme (eight colors or less) be used with this attribute to avoid overwhelming the color resources of the Genome Browser and your Internet browser.
blockCount - The number of blocks (exons) in the BED line.
blockSizes - A comma-separated list of the block sizes. The number of items in this list should correspond to blockCount.
blockStarts - A comma-separated list of block starts. All of the blockStart positions should be calculated relative to chromStart. The number of items in this list should correspond to blockCount.
参考BED文件格式
name- BED行名,在基因组浏览器左边显示;
score- 在基因组浏览器中显示的灰度设定,值介于0-1000;
strand- 正负链标记. Either "." (=no strand) or "+" or "-".
thickStart- feature起始位置(for example, the start codon in gene displays)。 When there is no thick part, thickStart and thickEnd are usually set to the chromStart position.
thickEnd- feature编码终止位置 (for example the stop codon in gene displays).
itemRgb- R,G,B (e.g. 255,0,0)值,当itemRgb设置为 "On",BED的行会显示颜色.
blockCount- blocks (exons外显子)数目.
blockSizes- blocks (exons)大小列表,逗号分隔,对应于blockCount.
blockStarts-blocks (exons)起始位置列表,逗号分隔,对应于blockCount.;这个起始位置是与chromStart的一个相对位置。

如果看到bed后面有数字,比如说BED3或者BED4,数字代表的可能是列数。
2.BED detail format
包含BED格式文件的4-12列,此外还有ID和a description of the item。
track name=HbVar type=bedDetail description="HbVar custom track" db=hg19 visibility=3 url="http://globin.bx.psu.edu/cgi-bin/hbvar/query_vars3?display_format=page&mode=output&id=$$"chr11 5246919 5246920 Hb_North_York 2619 Hemoglobin variantchr11 5255660 5255661 HBD c.1 G>A 2659 delta0 thalassemiachr11 5247945 5247946 Hb Sheffield 2672 Hemoglobin variantchr11 5255415 5255416 Hb A2-Lyon 2676 Hemoglobin variantchr11 5248234 5248235 Hb Aix-les-Bains 2677 Hemoglobin variant
粗体字必须包含在track-line里面。
3.BedGraph Track Format
track line attribute=value pairs
track lines define the display attributes for all lines in an annotation data set.
track line定义了注释文件集的展示属性。
name=<track_label>、description=<center_label>、type=<track_type> 、color=<RRR,GGG,BBB>等等
包含了四列BED文件的内容
Following the track definition line are the track data in four column BED format:

The chromosome coordinates are zero-based, half-open.
4.FASTA和FASTQ
1、FASTA文件的格式
在生物信息学中,FASTA格式(又称为Pearson格式)是一种基于文本的、用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来表示,且允许在序列前添加序列名及注释。
FASTA文件以序列表示和序列作为一个基本单元,各行记录信息如下:
第一行是由大于号">"开头的任意文字说明,用于序列标记,为了保证后续分析软件能够区分每条序列,单个序列的标识必须具有唯一性。;
从第二行开始为序列本身,只允许使用既定的核苷酸或氨基酸编码符号。通常核苷酸符号大小写均可,而氨基酸常用大写字母。使用时应注意有些程序对大小写有明确要求。文件每行的字母一般不应超过80个字符。
2、FASTQ文件格式
FASTQ是基于文本的、保存核酸序列和其测序质量信息的标准格式。其序列以及质量信息都是使用一个ASCII字符标示,最初由Sanger开发,目的是将FASTA序列与质量数据放到一起,目前已经成为高通量测序结果的事实标准。
FASTQ文件中以四行最为一个基本单元,并对应一条序列的测序信息,各行记录信息如下:
第一行记录序列标识以及相关的描述信息,以‘@’开头,为了保证后续分析软件能够区分每条序列,单个序列的标识必须具有唯一性;
第二行为碱基序列;
第三行以‘+’开头,后面是序列标示符、描述信息,或者什么也不加;
第四行,是质量信息,长度和第二行的序列相对应,每一个序列都有一个质量评分,根据评分体系的不同,每个字符的含义表示的数字也不相同。
5.BAM/SAM
BAM is the compressed binary version of the Sequence Alignment/Map (SAM) format, a compact and index-able representation of nucleotide sequence alignments. Many next-generation sequencing and analysis tools work with SAM/BAM.
SAM (Sequence Alignment/Map) format is a generic format for storing large nucleotide sequence alignments.
超链接可以查看以sam作为input或者output的软件。
处理大文件都需要一个索引,索引的作用是可以快速定位到文件的任意位置,因此,建立索引,也是bam文件的重要功能,而建立索引,必须是排序后的bam文件。所以,拿到一个比对好的sam之后,基本处理就是排序,格式转换,建立索引。
在bismark中有一个对基因组建立索引的步骤,但是我也不知道为什么要建立这个索引以及这个索引可以用来干什么。可能是为了提高比对的速度。留坑。
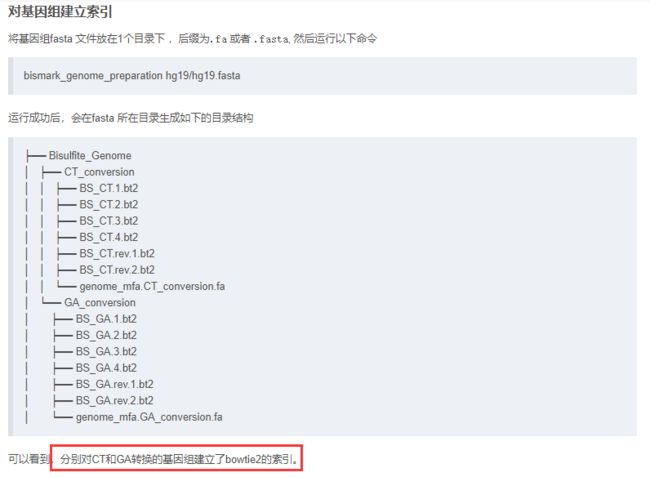
#这里的bt2结尾的文件就是索引,是二进制文件。
(1)Convert SAM to BAM using the samtools program:#将SAM文件转换为BAM文件。
samtools view -S -b -o my.bam my.sam
If converting a SAM file that does not have a proper header, the -t or -T option is necessary. For more information about the command, run samtools view with no other arguments.
(2)Sort and create an index for the BAM:排序并且建立索引
samtools sort my.bam my.sorted
samtools index my.sorted.bam
The sort command appends .bam to my.sorted, creating a BAM file of alignments ordered by leftmost position on the reference assembly.
The index command generates a new file, my.sorted.bam.bai, with which genomic coordinates can quickly be translated into file offsets in my.sorted.bam.有了这个.BAI为后缀的文件基因组坐标可以快速在BAM文件中转换为文件偏移量。(看不太懂)