GCE下载安装
在Linux系统下,直接在下载链接ftp中下载安装。
GCE主要包含两个主程序:kmer_freq_hash,用于k-mer频数统计;gce,用于基因组特征评估。
gce,基因组特征评估
可首先使用“gce -h”查看参数。
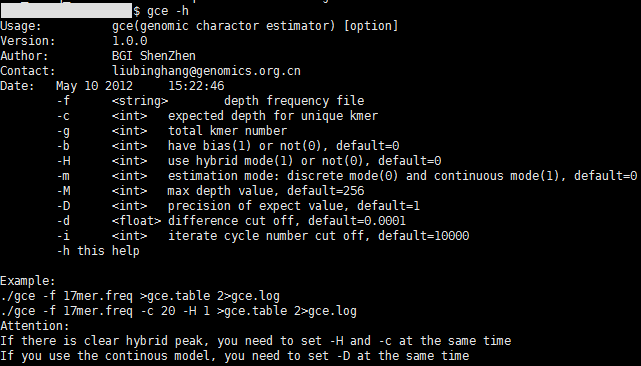
其中,常用参数选项说明:
-f,k-mer频数统计表;
-c,k-mer频数统计结果的“主峰”对应的频数,即k-mer曲线的主峰所对应的横坐标;
-g,k-mer片段总数,缺省时默认使用输入k-mer频数统计表文件计算k-mer总数;
-b,数据是(1)否(0,默认)有bias;
-H,使用杂合模式(1),或者单倍体模式(0,默认);
-m,估算模型的选择,离散型(0,默认)或连续型(1);
-M,设定计算时所支持的最大k-mer频数,默认256;
-D,期望值精度,默认为 1;若-m参数选择1,推荐该值设定为8;
非杂合模式示例
此处使用上述得到的k-mer频数统计结果文件“test.freq.stat”进行基因组特征评估。
输入文件“test.freq.stat”,该文件中,峰值对应k-mer频数等于58的位置,不再指定k-mer片段总数而是默认使用输入k-mer频数统计表文件计算k-mer总数,已知该物种基因组为单倍体,故不使用杂合模式,使用连续型估算模型且期望值精度设定为8,设置计算时所支持的最大k-mer频数256(若k-mer频数表由kmer_freq_hash得到,则可缺省,非kmer_freq_hash所得结果一定要设置该参数)。
程序运行完毕得到结果文件“test.gce.stat”和“test.gce.log”。
“test.gce.stat”为中间计算结果输出,一般情况下无需关注。
“test.gce.log”为程序运行的日志文件,同时记录了物种基因组特征评估结果。评估统计结果同样在该文件的最下方,

raw_peak,k-mer频数统计结果的“主峰”频数;
now_node,k-mer的种类数;
low_kmer,低覆盖度的k-mer数;
now_kmer,过滤低覆盖度的k-mer数后的k-mer总数;
cvg,估算出的测序平均深度;
genome_size,估算出的基因组大小,genome_size = now_kmer / cvg ;
a[1],仅出现1次的k-mer种类数占k-mer种类数总数的比值;
b[1],仅出现1次的k-mer片段数量占k-mer片段总数量的比值;该值可用于表征基因组中拷贝数为1的序列比例,则1-b[1]可认定为重复序列比例。
杂合模式示例
若测序物种的基因组高杂合(如通过k-mer曲线判断,例如曲线中存在杂合峰),我们可考虑运行杂合模式,此时相较于非杂合模式的运行结果会更佳。当然,若测序物种的基因组仅为简单基因组(无杂合),则是不能使用杂合模式运行的,强行运行杂合模式结果将相当不可靠。
此处继续使用上述得到的k-mer频数统计结果文件“test.freq.stat”进行基因组特征评估。虽然该测序物种的k-mer曲线显示其为单倍体,不可以使用杂合模式;但此处作为示例主要演示软件的使用,因此请无需在意该操作的正确性。
输入文件“test.freq.stat”,同上述,该文件中峰值对应k-mer频数等于58的位置,不再指定k-mer片段总数而是默认使用输入k-mer频数统计表文件计算k-mer总数,此处使用杂合模式,并使用连续型估算模型且期望值精度设定为8,设置计算时所支持的最大k-mer频数256(若k-mer频数表由kmer_freq_hash得到,则可缺省,非kmer_freq_hash所得结果一定要设置该参数)。
与上述结果一致,程序运行完毕得到结果文件“test.gce.stat”和“test.gce.log”。然后我们关注“test.gce.log”最下方的统计结果。

前面几项统计指标(raw_peak、now_node、genome_size等)是固定的,但统计结果与非杂合模式略有不同,需要注意。
此处多了杂合度评估结果,其中:
a[1/2],杂合k-mer种类数占k-mer种类数总数的比值;此时,kmer-species heterozygous ratio = a[1/2] / ( 2- a[1/2]),杂合率 = kmer-species heterozygous ratio / k-mer长度(例如本示例中为17);
b[1/2],杂合k-mer片段数量占k-mer片段总数量的比值;此时,1-b[1]-b[1/2]可认定为重复序列比例。
结合其它K-mer分析工具一起使用
由于GCE的kmer_freq_hash程序统计k-mer频数时,支持的最大频数深度为225,出现次数大于255的k-mer数量会与出现次数等于255的k-mer数量合并,因此有时可能无法满足分析需求。因此,我们可以考虑将GCE结合其它k-mer分析工具一起使用。
如下示例,依然使用了本次的测试数据。首先使用JELLYFISH进行k-mer频数统计,之后将JELLYFISH的结果输入至GCE中,评估物种基因组大小、重复序列含量等信息。由于JELLYFISH支持最大k-mer频数为10000,因此我们可知,结合JELLYFISH(JELLYFISH+gce)一起分析的结果与只使用GCE(kmer_freq_hash+gce)分析的结果相比会更加准确。
其中,JELLYFISH使用说明可参见:http://blog.sciencenet.cn/blog-3406804-1161522.html
基因组特征最终评估结果如下所示。特别注意,由于JELLYFISH支持最大k-mer频数为10000,因此在此处的gce参数中,-M设定为10001。
