TCGA学习01:数据下载与整理 -
TCGA学习02:差异分析 -
TCGA学习03:生存分析 -
TCGA学习04:建模预测-cox回归 -
TCGA学习04:建模预测-lasso回归 -
TCGA学习04:建模预测-随机森林&向量机 -
第二步:差异分析
前期文件
rm(list=ls())
load("gdc.Rdata")
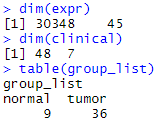
dim(expr)
dim(clinical)
table(group_list)
-
如前所述,这里使用老师的Rdata文件,存在tumor与normal的分组(但是矩阵与病人信息数量也不相同好像)
 gdc.Rdata
gdc.Rdata
预分析--PCA
- 观察组间差异是否明显
library("FactoMineR")#画主成分分析图需要加载这两个包
library("factoextra")
expr[1:4,1:4]
dat = log(expr+1) #防止为0
t(dat)[1:4,1:4] #行名为样本,列名为基因
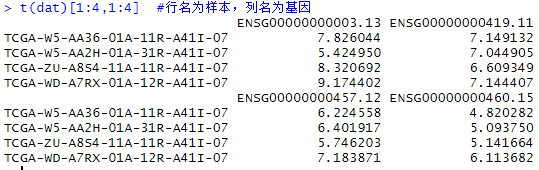
转置表达矩阵
dat.pca <- PCA(t(dat), graph = FALSE)
pca.plot=fviz_pca_ind(dat.pca, geom.ind = "point", col.ind = group_list,
addEllipses = TRUE, legend.title = "Groups")
pca.plot
-
如下图,可以看出两组差异还是比较明显的。
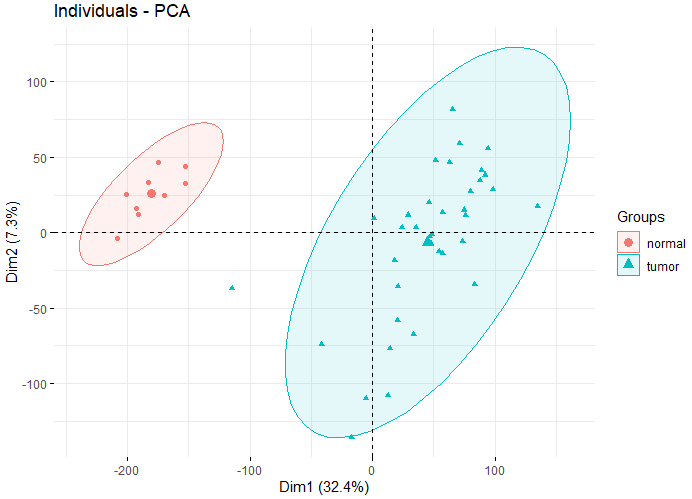 主成分分析图
主成分分析图
1、三种差异分析方法
(1) Deseq2
library(DESeq2)
colData <- data.frame(row.names =colnames(expr),
condition=group_list)
dds <- DESeqDataSetFromMatrix(
countData = expr,
colData = colData,
design = ~ condition)
dds <- DESeq(dds)
# 分组比较
res <- results(dds, contrast = c("condition",rev(levels(group_list)))) #rev设置下比较方式
resOrdered <- res[order(res$pvalue),] # 按照P值排序
DEG <- as.data.frame(resOrdered)
# 去除NA值
DEG <- na.omit(DEG)
head(DEG)
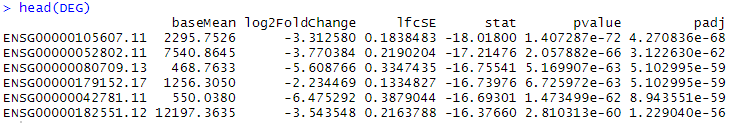
head(DEG)
#添加change列标记基因上调下调
logFC_cutoff <- with(DEG,mean(abs(log2FoldChange)) + 2*sd(abs(log2FoldChange)) )
#计算上下调标准
#logFC_cutoff <- 2
DEG$change = as.factor(
ifelse(DEG$pvalue < 0.05 & abs(DEG$log2FoldChange) > logFC_cutoff,
ifelse(DEG$log2FoldChange > logFC_cutoff ,'UP','DOWN'),'NOT')
)
head(DEG)
table(DEG$change)
DESeq2_DEG <- DEG
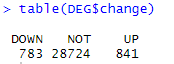
基因分为下调、无显著差异、上调三类
之前学习RNA-seq时,用的就是此方法。此外还有edgeR,limma两种方法,不太熟悉,仅记录下代码吧。会得到相似的三分类结果,但存在量的差异。
(2) edgeR
library(edgeR)
dge <- DGEList(counts=expr,group=group_list)
dge$samples$lib.size <- colSums(dge$counts)
dge <- calcNormFactors(dge)
design <- model.matrix(~0+group_list)
rownames(design)<-colnames(dge)
colnames(design)<-levels(group_list)
dge <- estimateGLMCommonDisp(dge,design)
dge <- estimateGLMTrendedDisp(dge, design)
dge <- estimateGLMTagwiseDisp(dge, design)
fit <- glmFit(dge, design)
fit2 <- glmLRT(fit, contrast=c(-1,1))
DEG=topTags(fit2, n=nrow(expr))
DEG=as.data.frame(DEG)
logFC_cutoff <- with(DEG,mean(abs(logFC)) + 2*sd(abs(logFC)) )
#logFC_cutoff <- 2
DEG$change = as.factor(
ifelse(DEG$PValue < 0.05 & abs(DEG$logFC) > logFC_cutoff,
ifelse(DEG$logFC > logFC_cutoff ,'UP','DOWN'),'NOT')
)
head(DEG)
table(DEG$change)
edgeR_DEG <- DEG
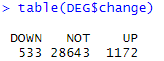
edgeR三分类
(3) limma
library(limma)
design <- model.matrix(~0+group_list)
colnames(design)=levels(group_list)
rownames(design)=colnames(expr)
dge <- DGEList(counts=expr)
dge <- calcNormFactors(dge)
logCPM <- cpm(dge, log=TRUE, prior.count=3)
v <- voom(dge,design, normalize="quantile")
fit <- lmFit(v, design)
constrasts = paste(rev(levels(group_list)),collapse = "-")
cont.matrix <- makeContrasts(contrasts=constrasts,levels = design)
fit2=contrasts.fit(fit,cont.matrix)
fit2=eBayes(fit2)
DEG = topTable(fit2, coef=constrasts, n=Inf)
DEG = na.omit(DEG)
logFC_cutoff <- with(DEG,mean(abs(logFC)) + 2*sd(abs(logFC)) )
#logFC_cutoff <- 2
DEG$change = as.factor(
ifelse(DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff,
ifelse(DEG$logFC > logFC_cutoff ,'UP','DOWN'),'NOT')
)
head(DEG)
table(DEG$change)
limma_voom_DEG <- DEG
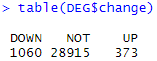
limma三分类
tj = data.frame(deseq2 = as.integer(table(DESeq2_DEG$change)),
edgeR = as.integer(table(edgeR_DEG$change)),
limma_voom = as.integer(table(limma_voom_DEG$change)),
row.names = c("down","not","up")
)
tj
save(DESeq2_DEG,edgeR_DEG,limma_voom_DEG,group_list,tj,file = "DEG.Rdata")
rm(list=ls())

tj
2、差异分析结果可视化
2.1 热图
- 挑选出显著差异基因
xz_DESeq2 = rownames(DESeq2_DEG)[DESeq2_DEG$change !="NOT"]
xz_DESeq2= dat[xz_DESeq2,]
xz_edgeR = rownames(edgeR_DEG)[DESeq2_DEG$change !="NOT"]
xz_edgeR= dat[xz_edgeR,]
xz_limma = rownames(limma_voom_DEG)[DESeq2_DEG$change !="NOT"]
xz_limma= dat[xz_limma,]
- 绘制热图
library(pheatmap)
library(ggplot2)
n=xz_DESeq2
# n=xz_edgeR
# n=xz_limma
n[1:4,1:4]
n = t(scale(t(n)))
n[1:4,1:4]
n_cutoff=2
#把绝对值对于2的count值改为2或者-2
n[n > n_cutoff] = n_cutoff
n[n < -n_cutoff] = -n_cutoff
n[1:4,1:4]
annotation_col = data.frame(group = group_list)
rownames(annotation_col) = colnames(n)
p.DEseq2 = as.ggplot(pheatmap(n, show_colnames = F, show_rownames = F,
annotation_col = annotation_col, legend = T, annotation_legend = T,
annotation_names_col = T))
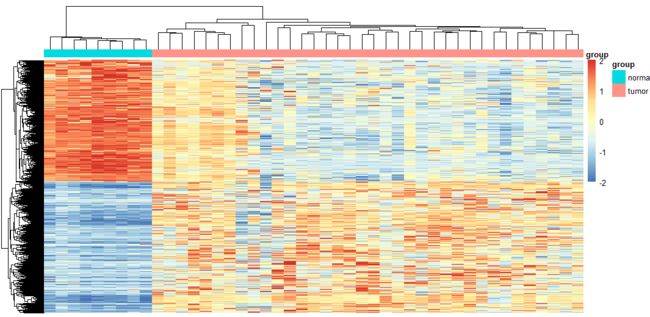
p.DEseq2
2.2 火山图
- 之前在差异分析是已经增加了down,not、up三分类变化的
change列,这里我们直接使用其来绘制
m2d = function(x){
mean(abs(x))+2*sd(abs(x))
}
dat=DESeq2_DEG
# dat=edgeR_DEG
# dat=limma_voom_DEG
logFC_cutoff=m2d(dat$log2FoldChange)
tile <- paste0("down gene:",
nrow(dat[dat$change == "DOWN",]),
"\n" ,
" up gene:" ,
nrow(dat[dat$change == "UP",]) )
ggplot(data = dat, aes(x = log2FoldChange, y = -log10(pvalue))) +
geom_point(alpha = 0.4, size = 3.5, aes(color = change)) +
scale_color_manual(values = c("blue", "grey", "red")) +
geom_vline(xintercept = c(-logFC_cutoff, logFC_cutoff), lty = 4, col = "black", lwd = 0.8) +
geom_hline(yintercept = -log10(0.05), lty = 4,col = "black", lwd = 0.8) +
theme_bw() +
labs(title = tile, x = "logFC", y = "-log10(P.value)") +
theme(plot.title = element_text(hjust = 0.5))
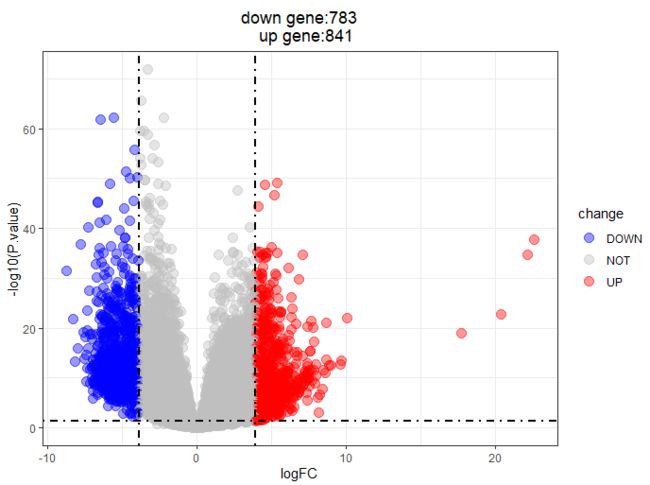
DESeq2_DEG火山图
- 可以利用patch包将上面的六张图拼在一起,类似如下代码
library(patchwork)
(h1 + h2 + h3) / (v1 + v2 + v3) +plot_layout(guides = 'collect')
3、三种差异分析方法比较
3.1 韦恩图展示三方法的三分类差异
#定义取类函数
UP=function(df){
rownames(df)[df$change=="UP"]
}
DOWN=function(df){
rownames(df)[df$change=="DOWN"]
}
#画韦恩图
ven.plot=venn.diagram(list(Deseq2 = UP(DESeq2_DEG), edgeR = UP(edgeR_DEG), limma = UP(limma_voom_DEG)),
imagetype = "png", filename = NULL,
lwd = 1, lty = 1,
col = c("#0099CC", "#FF6666", "#FFCC99"),
fill = c("#0099CC", "#FF6666","#FFCC99"),
cat.col = c("#0099CC", "#FF6666", "#FFCC99"),
cat.cex = 1.5, rotation.degree = 0,
main = "name", main.cex = 1.5, cex = 1.5, alpha = 0.5,
reverse = TRUE)
#结果返回是一个list,看不太懂,反正不是图..看了下老师教程,加了如下代码就可以了。
#猜测是不同绘图系统导致的吧?
library(cowplot)
p.up = as.ggplot(as_grob(ven.plot))
# 同法也可绘制出 p.down
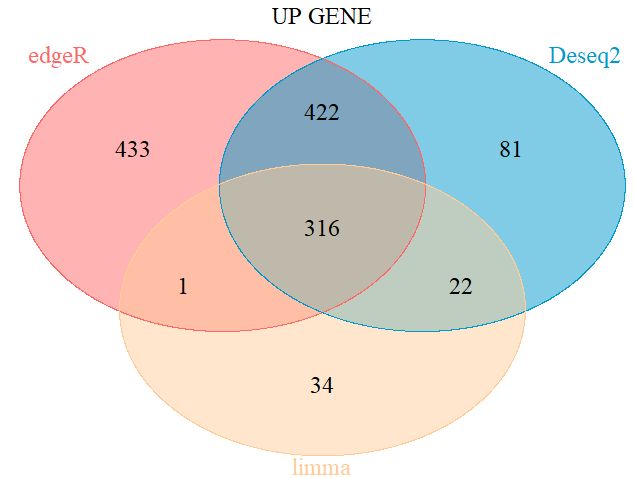
三种方法韦恩图,316个相同
3.2 用三方法相同差异基因绘制热图
#挑选出共同基因
up = intersect(intersect(UP(DESeq2_DEG),UP(edgeR_DEG)),UP(limma_voom_DEG))
down = intersect(intersect(DOWN(DESeq2_DEG),DOWN(edgeR_DEG)),DOWN(limma_voom_DEG))
#套入绘图流程
n=expr[c(up,down),]
# n=xz_edgeR
# n=xz_limma
n[1:4,1:4]
n = t(scale(t(n)))
n[1:4,1:4]
n_cutoff=2
#把绝对值对于2的count值改为2或者-2
n[n > n_cutoff] = n_cutoff
n[n < -n_cutoff] = -n_cutoff
n[1:4,1:4]
annotation_col = data.frame(group = group_list)
rownames(annotation_col) = colnames(n)
hp = as.ggplot(pheatmap(n, show_colnames = F, show_rownames = F,
annotation_col = annotation_col, legend = T, annotation_legend = T,
annotation_names_col = T))
#如下图,差异很明显
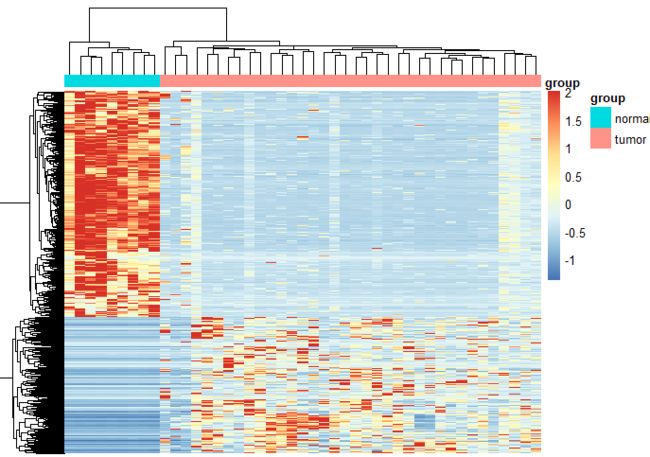
共有基因热图
3.3 最后拼一张综合图收尾吧
library(patchwork)
pca.plot + hp + up.plot + down.plot
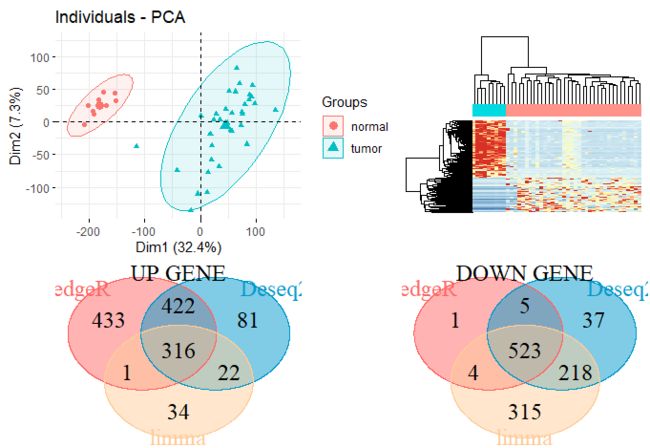
PCA+heatmap+veen