Seurat使用教程(v3.0)
Seurat是一个分析单细胞转录组数据的R包,用于QC,分析和探索单细胞RNA-seq数据,相关资料如下:
参考教程
测试数据
具体流程如下:
graph TD
A[安装Seurat包] -->|加载Seurat包| B(读入10X数据)
B --> C(QC及前处理)
C --> |筛选出符合要求的数据| D(标准化数据)
D --> E(PCA分析)
E--> F(t-SNE降维分析或者UMAP降维分析)
F -->G(细胞聚类)
Seurat包安装
在linux或者windows中的Rstudio均可以安装运行,与安装其他R包一致。
install.packages("Seurat")
install.packages("dplyr")
数据导入
library(Seurat)
library(dplyr)
cip <- readRDS("subset_cip.rds")
cip1 <-CreateSeuratObject(counts = cip,project = "CIPs",min.cells = 3,min.features = 200)
cip1[["percent.mt"]] <- PercentageFeatureSet(object = cip1, pattern = "^mt-")
head(x = [email protected], 5)
pbmc
结果如下
orig.ident nCount_RNA nFeature_RNA percent.mt
TTAGTTCAGCTGATAA-1 CIPs 23206 4473
3.412910
CTAGCCTAGAGACGAA-1 CIPs 9548 2803
4.733976
GTGAAGGGTCTAAAGA-1 CIPs 12292 3179
4.905630
CATCCACTCAATAAGG-1 CIPs 14012 3501
5.338281
TTCCCAGCACTGCCAG-1 CIPs 14704 3604
5.597116
QC及预处理
预处理该步骤非必须,可根据实际情况作出相应变动;其目的就是根据基因的表达量、细胞中及线粒体基因表达量等特征,对细胞进行一个初步的过滤,就是根据可视化结果指定一个阈值,达到或者超过设定的阈值才进入到下游后续分析,阈值设定常用的几个指标:nGene、nUMI、mito.percent
VlnPlot(object = cip1, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),ncol = 3)

1.png
根据图片选定阈值排除一些离群值后
plot1 <- FeatureScatter(object = cip1, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(object = cip1, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
CombinePlots(plots = list(plot1, plot2))
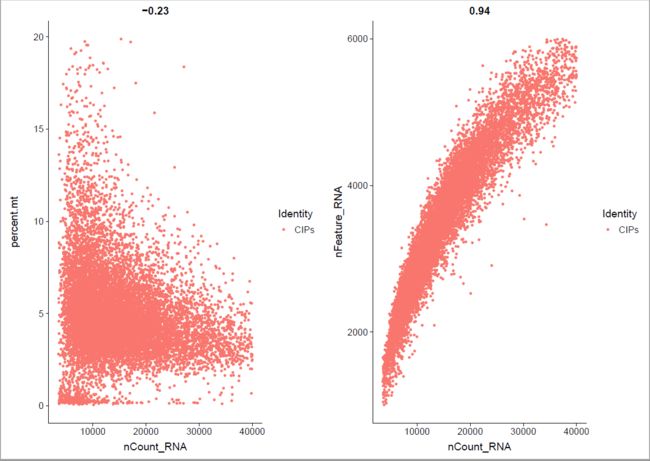
2.png
标准化处理
归一化
cip1 <- subset(x = cip1, subset = nFeature_RNA > 1000 & nFeature_RNA < 5800 & percent.mt < 13)
cip1 <- NormalizeData(object = cip1, normalization.method = "LogNormalize", scale.factor = 10000)
缩放数据及线性降维
all.genes <- rownames(x = cip1)
cip2<- ScaleData(object =cip1, features = all.genes)
cip2<- RunPCA(object = cip2, features = VariableFeatures(object = cip2))
VizDimLoadings(object = cip2, dims = 1:2, reduction = "pca")
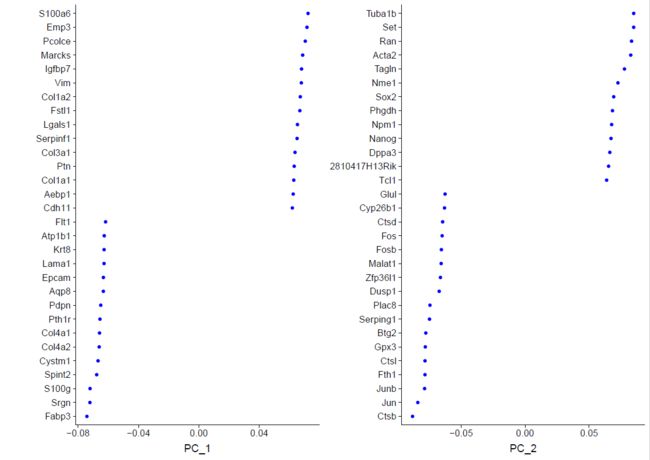
3.png
热图绘制
DimHeatmap(object = cip2, dims = 1, cells = 500, balanced = TRUE)
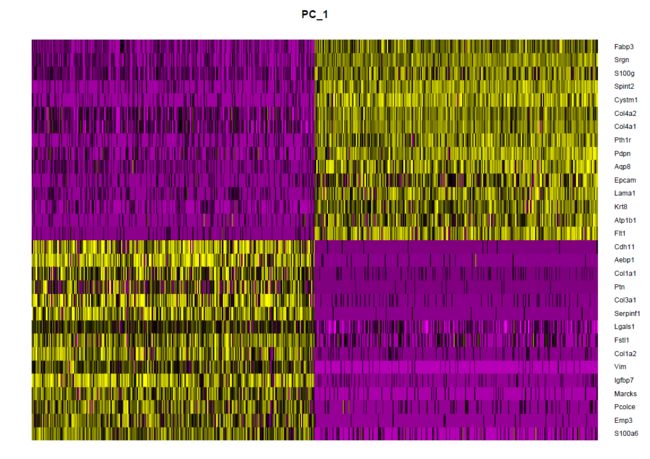
4.png
DimHeatmap(object = cip2, dims = 1:15, cells = 500, balanced = TRUE)

5.png
维数选择
cip2 <- JackStraw(object = cip2, num.replicate = 100)
cip2 <- ScoreJackStraw(object = cip2, dims = 1:20)
JackStrawPlot(object = cip2, dims = 1:15)
ElbowPlot(object = pbmc)
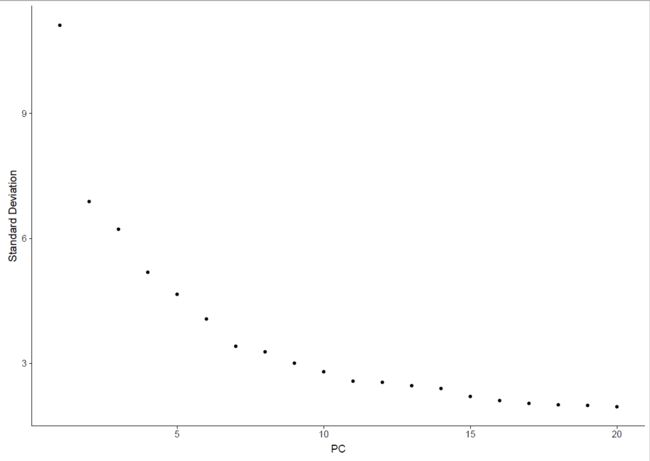
6.png
细胞聚类
cip2 <- FindNeighbors(object = cip2, dims = 1:10)
cip2 <- FindClusters(object = cip2, resolution = 0.5)
head(x = Idents(object = cip2), 5)
t-SNE可视化结果
cip2 <- RunTSNE(object = cip2, dims = 1:10)TSNEPlot(object = cip2)

7.png
具体说明后续继续补充。