参考: 从零开始完整学习全基因组测序数据分析:第3节 数据质控
前言
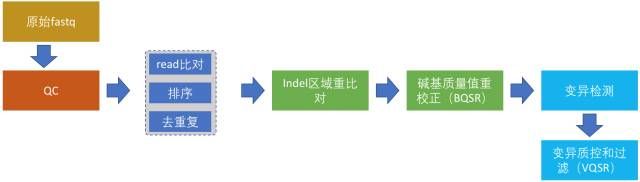
如上图显示,整个完整的WGS 流程一般有以下步骤:
1)获取测序数据。
2)原始测序数据的质控。
3)read比对,排序,排除重复序列。
4)index 区域重新比对。
5)碱基质量值重新校正。
6)变异检测。
7)变异结果质控和过滤。
数据质控
数据质控的意义
由测序相关的内容可知,目前主流的测序手段还是以illumina 为首的二代测序技术。
而边合成边测序,碱基的合成,依靠于DNA聚合酶。而随着合成链的不断延伸,DNA聚合酶的效率则不断下降,而特异性也会不断变差。
这也导致了二代测序的明显缺点:越到后面碱基合成的错误率越高。
正因此一般来说市面的NGS 测序手段都是小片段的读取。
除了测序的后期,由于测序仪在开始工作时也由于反应不够稳定,同样会带来质量值的变动。
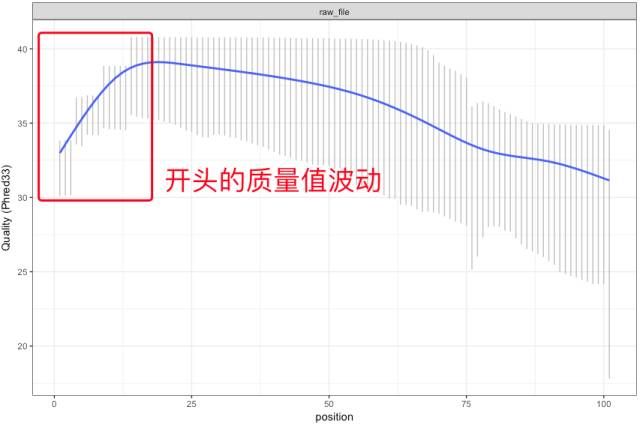
分析测序数据
测序数据的好坏直接影响到我们的下游分析。而不同的测序平台其错误率的图谱都是存在差异的。
因此,在分析测序数据前非常需要做到两个方面:1)原始数据的来源;2)评估对于分析的影响。
这是我们认识数据质量的第一步。了解原始数据通过何种测序平台产生,以及错误率分布、偏向性与局限性、是否会显著受GC含量的影响等。
除了查看使用的测序手段的相关资料外,最好的做法便是自己分析。
认识你的数据,不要相信你的工具。
学会认识测序数据
一般可以从下面几个方面来分析。

read各个位置的碱基质量值分布
通过FASTQ 的学习,我们知道,通过shell 脚本判断,以及通过设计简单的python 代码,可以方便我们实现质量值由ASCII 码到数值的转换。
[35, 20, 17, 18, 24, 34, 35, 35, 35, 34, 35, 34, 29, 29, 32, 32, 34, 34, 33,
29, 33, 33, 32, 35, 35, 35, 34, 34, 34, 34, 35, 35, 34, 35, 34, 35, 34, 35,
34, 34, 34, 35, 35, 35, 35, 34, 33, 33, 30, 33, 24, 27]
但很显然,输入以上这般的列表,对于成千上万个碱基数目来说,显然不够直观,需要更加直观的方式——画图。
没错,就像最开始的那副图一般,通过线图的方式,将质量值的变化趋势直观的表现出来。
市面上最常用的软件之一,便是FastQC。它是一个java 程序,可以同时给出多个方面的数据图,包括测序数据的QC报告等。
在做read 质量值分析时,FastQC并不单独查看具体的每条read中碱基的质量值,而是将FASTQ 文件中的所有read 数据综合到一起分析。
下图便是一个质量非常好的read 各位置碱基质量值分布。
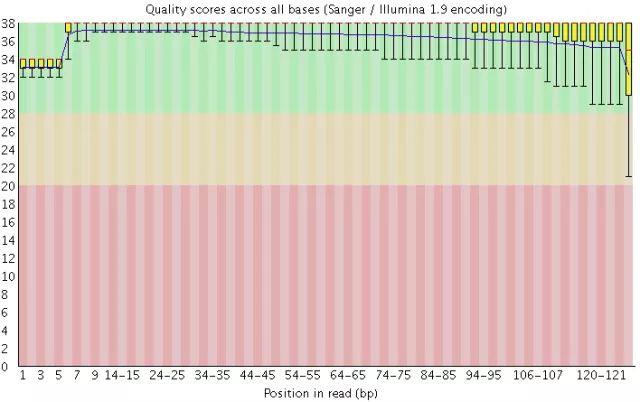
如何来解读这张图呢?
这张图的横轴是read 的碱基位置,纵轴为碱基质量值。在这个例子里,read的长度为126bp(HiSeq X10测序)。
read 上每一个位置都有一个箱型图,表示在该位置上,所有碱基的质量分布。由上图可知,除了最后一个碱基外,其他的碱基质量值均值都在30以上,且波动很小。(绿色背景表示高质量区域)
坏的测序结果也可能会很可怕
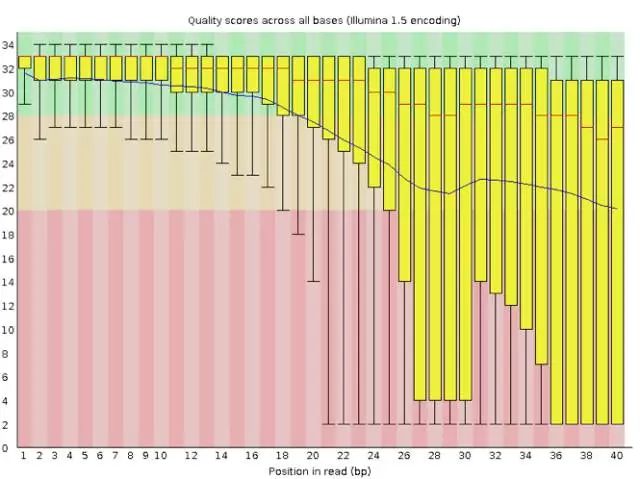
不难看出,该read 各个位置质量值不仅波动大,而且很多read 的值甚至跌落到红色区域。
最好的情况是重新测序,但如果不得不使用这个数据,就要把这些低质量的数据全都去除掉才行,同时还需留意是否还存在其他的问题,但不管如何都一定会丢掉很大一部分的数据。
碱基总体质量值分布
除了各个read 值的分布外,FastQC还提供了碱基总体质量值分布。
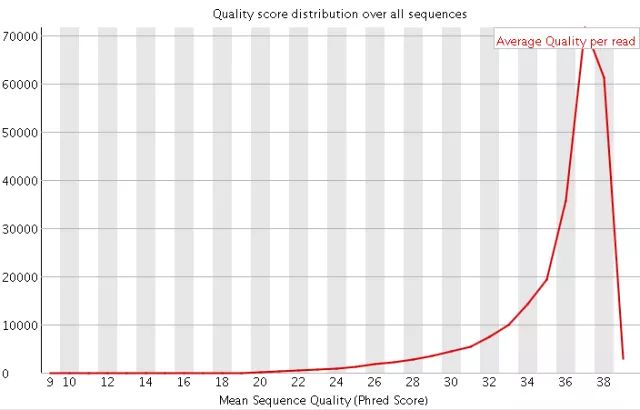
一般来说,Q20与Q30 是衡量碱基质量值的关键指标。一般来说,对于二代测序,要求达到Q20 碱基要在95%以上(不能低于90%),而Q30 碱基要在85%以上(不能低于80%)。
read 各个位置碱基分布比例
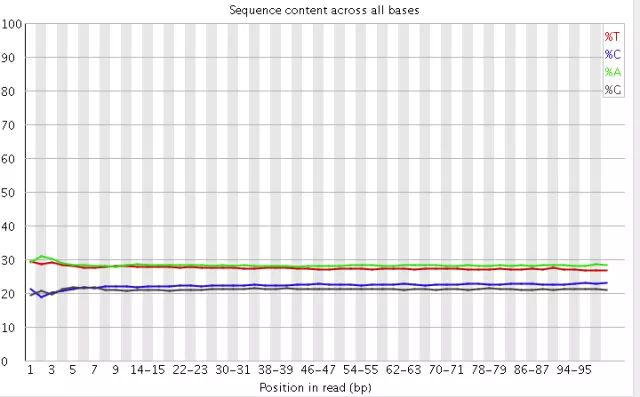
了解read 各个位置碱基分布比例,目的是为了分析碱基的分离程度。
什么是碱基分离?
一般来说,碱基遵循AT、CG的配对原则,若测序过程是随机的话,则每个位置上的A与T,C与G 的比例应该是接近的。
一般来说,最好平均值偏差在1%以内,除非有合理原因(某些特定的测序需求)。
GC 含量分布图
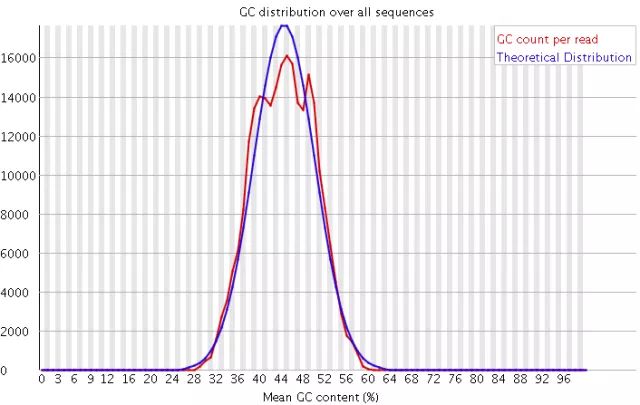
GC 含量是指 G、C 碱基数量占总碱基数量的比例。
二代测序平台会存在一定的测序偏向性,而我们可以通过GC 含量值来判定测序过程是否足够随机。
对于人类来说,基因组的GC% 一般在40%左右。
因此通过比较分布图上的GC分布与理论值的拟合,如果发现GC含量的图谱明显偏离这个值,则说明测序过程存在比较高的序列偏向性。
说明可能基因组中的某些特定序列被反复测序的几率高于平均水平,除了覆盖度会有偏离之后,将会影响下游的变异检测和CNV分析。
N含量分布图
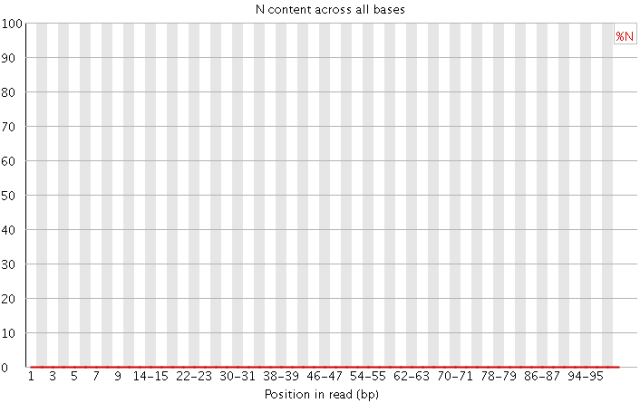
一般来说,N在测序中不应该出现。如果出现则意味着测序的光信号无法被清楚的解析。若该情况发生较多的话,往往意味着测序系统或者测序试剂的错误。
read 中有无接头序列
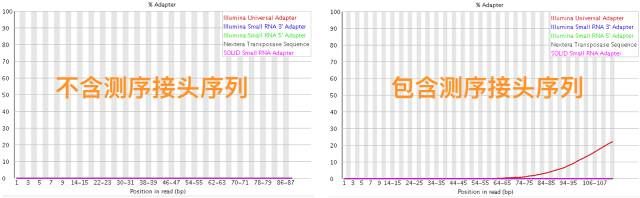
在二代测序中提到,在构建DNA文库时,被打散的短的DNA 链会在两头修饰,加上一段接头,用于互补结合到flowcell 上,以及用于区分这些DNA 序列的来源。
当测序read 的长度大于被测序的DNA片段时,就会在read 的末尾检测到这些接头序列。
一般来说,WGS 测序都不会检测到接头序列,因为构建WGS 的文库序列一般都在几百bp,而read 的测序长度一般只有100-150bp。
但有时候在进行RNA 测序时,由于有的RNA 序列比较短(几十bp),则会比较容易出现read 中出现接头序列的情况。
FastQC 使用
可以到官网进行下载
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
也可以通过命令行 使用 wget 命令。
wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.5.zip ./
这里推荐一个Linux 指令tree,方便查看文件结构。
小插曲 安装tree和java
参考 https://blog.csdn.net/etalien_/article/details/86361260
使用wget 下载
$ wget http://mama.indstate.edu/users/ice/tree/src/tree-1.7.0.tgz
解压
$ tar -zxvf tree-1.7.0.tgz
进入目录
$ cd tree-1.7.0
make tree
$ make
cd tree到bin目录
$ cp tree /bin
这时候我们顺便再安装一下java
参考:https://blog.csdn.net/lyhkmm/article/details/79524712
到官网找到对应安装包
下载
wget https://download.oracle.com/otn/java/jdk/8u251-b08/3d5a2bb8f8d4428bbe94aed7ec7ae784/jdk-8u251-linux-x64.tar.gz?AuthParam=1589801751_99c6e599266b8b501cc9a981b68aa5ff
创建java 文件目录
$ mkdir -p /usr/local/java
解压到文件目录
$ tar -zxvf java.tar.gz -C /usr/local/java/
添加环境变量
$ vi /etc/profile
'''
在文件中加入以下环境信息
我的java 安装版本为jdk1.8.0_251
export JAVA_HOME=/usr/local/java/jdk1.8.0_251
export CLASSPATH=$CLASSPATH:$JAVA_HOME/lib/
export PATH=$PATH:$JAVA_HOME/bin
'''
重新加载配置文件
$ source /etc/profile
测试一下,成功则可以看到java 版本信息。
$ java -version
继续之前
安装完成后,进行解压就可以使用
unzip fastqc_v0.11.5.zip
再修改以下文件夹中fastqc的权限。
chmod 755 fastqc
# 这里我也不是太懂
fastqc 使用
通过-o 参数可指定FastQC 报告的输出目录。默认下,FastQC结果会输出到测序文件untreated.fq 同一目录下。
$ /path_to_fastqc/FastQC/fastqc untreated.fq -o fastqc_out_dir/
fastqc 的输出文件只有两个,一个.html和一个.zip。
除此之外,还可以同时输入多个.fq 文件。
$ /path_to_fastqc/FastQC/fastqc /path_to_fq/*.fq -o fastqc_out_dir/
除了Linux外,FastQC 也提供图形化软件。

处理序列文件
由之前内容知,测序文件中可能会存在接头序列,以及read 低质量的序列,面对这些有时候不得不要对这些序列进行进一步处理。
除了自己编写一些代码以进行个性化的fq 数据过滤,还有一些现成的工具,如SOAPnuke、cutadapt、untrimmed等。还有Trimmomatic(也是一个java程序)、sickle和seqtk等比较好用的程序。
其中Trimmomatic 可以用来切除illumina 测序平台的接头序列,而且还可以去除指定的接头序列以及过滤read 末尾的低质量碱基序列。
然而sickle和seqtk,只能去除低质量的碱基。
其具体的原理是通过滑动一定长度的窗口,接着计算窗口内的碱基平均质量。如果质量过低,就直接往后全部切除。
直接全部从某个起始位置全部切除。

由上可知,如果获得的fq数据中不含有这些测序接头,那么除了trimmomatic外,也可以直接使用sickle 与seqtk,而后面这两个软件处理的更快,消耗的资源也更少。
使用trimmomatic 构造序列过滤流程
首先在官网下载
http://www.usadellab.org/cms/?page=trimmomatic
还是用wget下载,然后便可以进行解压。获得一个java 可执行文件。
$ wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.39.zip
$ unzip Trimmomatic-0.39.zip
通过tree 命令可以看到其结构
完成后可以直接使用java 运行它。
序列文件的选择
adapters 是Trimmomatic 中非常重要的部分。
其中存放的是illumina 测序平台的接头序列(fasta序列),在实际使用中,如果需要切除头序列,则需要明确指定对应的序列作为输入参数。
那么该如何选择接头序列呢?目前的HiSeq 与MiSeq 系列使用的都是TruSeq3(上图tree 中)。而TruSeq2则是先前的GA2系列测序仪使用的,已经很少见了。
即使用的不是illumina 的测序平台, 也可以仿照Trimmomatic 中的这些adapter 文件夹下的这些文件的格式做一个新的接头序列,然后再作为参数传入。在自定义接头文件时,还需要注意一下命名的细节。
由tree 中的adapters 文件名称也能看出,Trimmomatic 有两种运行模式:PE 与SE。
而具体使用PE(pair end) 还是SE(single end)就按照具体的测序类型进行选择。
PE 模式
$ java -jar /path/Trimmomatic/trimmomatic-0.39.jar PE -phred33 -trimlog logfile reads_1.fq.gz reads_2.fq.gz out.read_1.fq.gz out.trim.read_1.fq.gz out.read_2.fq.gz out.trim.read_2.fq.gz ILLUMINACLIP:/path/Trimmomatic/adapters/TruSeq3-PE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50
SE 模式
java -jar /path/Trimmomatic/trimmomatic-0.39.jar SE -phred33 -trimlog se.logfile raw_data/untreated.fq out.untreated.fq.gz ILLUMINACLIP:/path/Trimmomatic/adapters/TruSeq3-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50
在之前的FASTA 与FASTQ 中介绍过,质量数一般有三种体系:1)Phred33;2)Phred64;3)Solexa64。
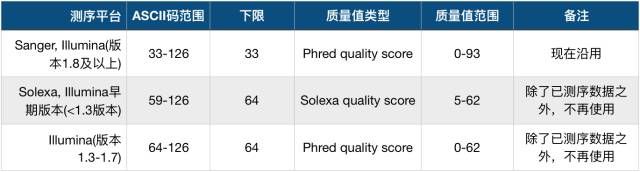
不同的质量数体系对应不同的ASCII码解析方式,也就对应着不同的质量数的计算方式。但Trimmomatic 默认采用Phred64,但目前主流的体系已经变成Phred33,所以使用时需要注意调整。
输入输出文件
通过输入
$ java -jar trimmomatic-0.39.jar
可以了解SE 与PE 用法的详情。
Usage:
# PE 用法
PE [-version] [-threads ] [-phred33|-phred64] [-trimlog ] [-summary ] [-quiet] [-validatePairs] [-basein | ] [-baseout | ] ...
or:
# SE 用法
SE [-version] [-threads ] [-phred33|-phred64] [-trimlog ] [-summary ] [-quiet] ...
不难看出-basein 与-baseout 中的basein 与baseout 可以省略。
除此之外,相比SE 模式,PE 模式的测序过程,需要指定一个文件夹用于存放被过滤掉的read 信息out.trim.read_2.fq.gz。
在PE 与SE 模式的最后,还有一个重要的点,就是< Trimmer >参数的设定。该参数包括很多切除接头序列和低质量序列的细节。
1)ILLUMINACLIP:接头序列切除参数。比如TruSeq3-PE.fa:2:30:10就分别代表了TruSeq3-PE.fa是接头序列,而比对时接头序列时允许最大的错误数为2;30 指PE 的两条read 同时和PE 的adapter 序列比对时,匹配度如果超过30%,则认为该PE 的read 中包含adapter,需要被切除。而最后的10则表示,只要这条read的某部分与adpater序列有超过10%的匹配率,就代表含有adapter。
2)SLIDINGWINDOW:滑动窗口参数。SLIDINGWINDOW:5:20代表窗口长度为5,窗口中的平均质量数至少为20,否则会切除;
3)LEADING:LEADING:5规定read 开头碱基切除的阈值。
4)TRAILING:TRAILING:5规定read 末尾碱基切除的阈值。
5)MINLEN:规定read被切除后至少保留的碱基长度,如果低于该长度,则会被丢弃。
除此之外,Trimmomatic一般报错难以查询问题,但主要问题还是参数或者是语法使用的问题。