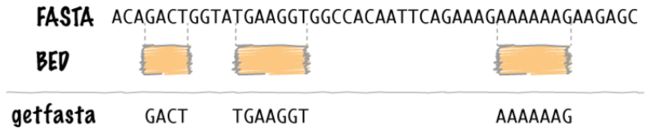
getfasta可以根据BED/GFF/VCF文件提供的feature在染色体上的位置信息,从fasta中提取feature的碱基序列
bedtools getfasta [OPTIONS] -fi
-bed -fo
Tip
- The headers in the input FASTA file must exactly match the chromosome column in the BED file.#FASTA 中序列名与BED文件中染色体名字要一一对应,不然找不到;类似于perl中hash,python中字典。
- You can use the UNIX
foldcommand to set the line width of the FASTA output. For example,fold -w 60will make each line of the FASTA file have at most 60 nucleotides for easy viewing. - BED files containing a single region require a newline character at the end of the line, otherwise a blank output file is produced.
$ bedtools getfasta -h
Tool: bedtools getfasta (aka fastaFromBed)
Version: v2.25.0
Summary: Extract DNA sequences into a fasta file based on feature coordinates.
Usage: bedtools getfasta [OPTIONS] -fi -bed -fo
Options:
-fi Input FASTA file #samtools先建立index再使用
-bed BED/GFF/VCF file of ranges to extract from -fi
-fo Output file(can be FASTA or TAB-delimited)
-name Use the name field for the FASTA header
-split given BED12 fmt., extract and concatenate the sequencesfrom the BED "blocks" (e.g., exons)
-tab Write output in TAB delimited format.#格式:name \t sequence
- Default is FASTA format.
-s Force strandedness. If the feature occupies the antisense strand, the sequence will be reverse complemented.- By default, strand information is ignored.#考虑链的正负方向,+提取正链序列,-提取正链序列
-fullHeader Use full fasta header.- By default, only the word before the first space or tab is used.
例子:
结果文件中fasta格式序列名字默认格式: “-name可设定提取的序列名字为对应的BED文件中feature名字
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10
$ bedtools getfasta -fi test.fa -bed test.bed
>chr1:5-10
AAACC
# optionally write to an output file
$ bedtools getfasta -fi test.fa -bed test.bed -fo test.fa.out
$ cat test.fa.out
>chr1:5-10
AAACC
#-name设定提取的序列名字为对应的BED文件中feature名字
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10 myseq
$ bedtools getfasta -fi test.fa -bed test.bed -name
>myseq
AAACC
-s #考虑链的正负方向,+提取正链序列,-提取正链序列
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 20 25 forward 1 +
chr1 20 25 reverse 1 -
$ bedtools getfasta -fi test.fa -bed test.bed -s -name
>forward
CGCTA
>reverse
TAGCG