炎症性肠病(IBD)是一种慢性且易复发的肠道炎症性疾病。全球500多万人受其困扰。

这种疾病是由多种遗传和环境因素共同作用引起的,这些因素改变了肠道的稳态,从而引发了基因易感个体的免疫介导炎症反应。
炎症性肠病包括克罗恩病和溃疡性结肠炎。
两者异同:
虽然克罗恩病和溃疡性结肠炎的发病机制都涉及肠道炎症,但这两种疾病在几个方面有所不同,包括与特定易感位点的关系、与疾病相关的免疫反应和病理类型。

这两种形式的IBD发病机制的一个关键方面是它们与生活在肠道中的共生微生物的存在有关。
寄主共生体参与了许多宿主的生理过程,包括消化和代谢功能、上皮屏障的调节、宿主免疫系统的发育和调节以及对病原体定植的保护。
IBD的一个关键特征是肠道微生物群失调,然而,失调在疾病中的确切作用仍然鲜为人知。

大量共生微生物靠近上皮表面是对粘膜免疫系统的一个独特挑战,因为它必须保持对入侵病原体的免疫反应的能力,同时避免对肠道微生物群产生有害的炎症反应。
这篇文章主要围绕以下几个问题展开讨论:
被称为“粘膜防火墙”的保护机制包含哪几种策略来维持稳态?
IBD相关的基因突变如何破坏粘膜防火墙?
IBD相关的遗传缺陷如何导致病变积累和渗透到肠道组织,从而进一步促进失调和炎症?
如何将微生物群作为IBD的潜在治疗方法?
粘膜防火墙
肠道中大量的共生微生物被单层上皮细胞从宿主组织中分离出来。为了维持其与肠道菌群的稳态关系,宿主已经进化出几种策略来减少微生物与上皮表面的接触,并限制可能引发不必要的炎症反应的渗透共生体的存在。
促进共生体与上皮分离的保护机制
宿主通过几种机制限制潜在有害的微生物接近粘膜表面,包括粘液分泌、抗菌蛋白和免疫球蛋白释放到肠腔。
我们先结合图1来看看大小肠的特点。
在大肠中,有两个明显不同的粘液层:
而小肠呢,没有像大肠那样清晰的内部粘液层,但它含有大量的Paneth细胞,这是一种特殊的肠道细胞,富含抗菌分子。为响应细菌刺激,Paneth细胞将富含的抗菌分子(包括α-防御素)释放到肠腔中,以限制共生菌的积累。
此外,小肠中的肠上皮细胞(IEC)分泌抗微生物凝集素,例如再生的胰岛衍生蛋白3γ(REG3γ),其在粘液层中积累并进一步促进微生物从宿主中的分离。
【名词小讲堂】
肠上皮细胞(IEC):
· 肠道上皮细胞是体内与外界之间的一个重要的保护屏障。
· 它由不同功能性的肠道干细胞群体共同维持,是具有极性的柱状上皮细胞,参与肠道的消化,吸收,分泌,免疫屏障和应激反应等。
· 粘膜上皮内含有大量的免疫细胞和免疫分子,是机体内最大的免疫组织。
再来看固有层(也就是图1 蓝色部分)
虽然固有层基本不存在急性炎症细胞,但某些嗜中粒细胞可以稳定迁移至肠腔侧,通过多种机制杀死上皮表面附近的细菌,包括诱导氧化猝发。
【名词小讲堂】
氧化猝发(Oxidative burst ):可直接杀死细菌的吞噬细胞迅速产生活性氧。
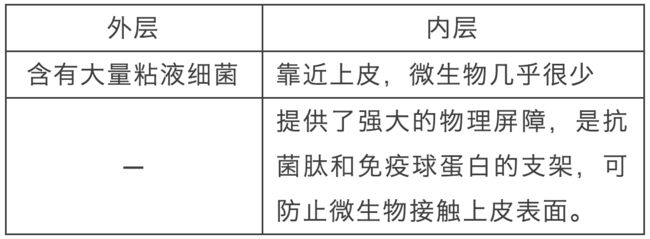
图1 粘膜防火墙
图注:杯状细胞分泌糖蛋白,包括粘蛋白2(MUC2),形成粘液屏障,防止肠腔中的微生物与上皮接触。在小肠中,松散形成的粘液层包裹上皮细胞,而在大肠中,内部粘液层基本上没有细菌,粘液共生体仅限于外部粘液层。
位于小肠隐窝的Paneth细胞构成性地表达杀微生物α-防御素。微折叠(M)细胞是一种特殊的上皮细胞,覆盖淋巴组织,如Peyer‘s斑片,便于抗原取样。树突状细胞(DCS)样本抗原在Peyer‘s斑片中由m细胞传递,或可能直接在腔内通过投射穿透上皮层。
分泌性IgA(SIgA)是由位于 Peyer‘s 斑片中的浆细胞产生的,通过转胞分泌穿过上皮进入肠腔。大多数 SIgA 是在 Toll 样受体(TLR)通过微生物抗原参与T细胞和 b 细胞后产生的,并与广泛的肠道微生物低亲和力结合,防止它们在上皮中易位。滤泡辅助(TFH)细胞依赖性反应通过支持高亲和力IgA的产生进一步促进稳态,后者与选择细菌结合。
髓系细胞衍生细胞因子,包括IL-1β、IL-6和IL-23,促进t辅助17(Th17)细胞分化和第3组固有淋巴样细胞(ILC3)的激活。由ILC3S和Th17细胞产生的IL-22通过诱导抗菌肽(AMPS)的表达,包括再生胰岛衍生蛋白3γ(Reg3γ),进一步加强共生体的分离。
il-22还能促进岩藻糖基转移酶2(fut2)介导的上皮性糖聚糖的岩藻糖基化,以支持微生物共生,防止潜在有害细菌的入侵。如果细菌能够克服这些障碍,IL-17刺激的中性粒细胞可以消除根尖上皮表面附近的病理组织或那些已经到达固有层的病理组织。
TH1细胞源性干扰素γ(IFNγ)进一步增强了巨噬细胞和DCs对微生物的杀灭作用。细菌特异性IgG的产生通过结合病理生物促进调理作用来控制全身感染。此外,渗透微生物被肝和脾巨噬细胞中的Kupffer细胞吞噬,以控制系统传播。
接着看下细胞因子IL-22的作用。
细胞因子IL-22还通过作用于上皮细胞介导屏障功能和抗微生物宿主防御,在建立宿主-微生物相互作用中发挥作用。例如,第3组固有淋巴细胞(ILC3s)分泌IL-22是抑制共生细菌所必需的,因为它诱导了抗菌肽的表达,从而阻止了Alcaligenes spp 的全身性传播。
IL-22的天然来源对于控制肠道内分段丝状菌(SFB)的增殖和限制T辅助17(TH17)细胞介导的结肠炎也很重要。
【名词小讲堂】
分段丝状菌(SFB),一种与梭菌有关的细菌,主要居住在小鼠的末端回肠,并促进辅助17细胞的发育。
IL-22还促进上皮聚糖岩藻糖基化,以支持适应岩藻糖作为营养源的共生细菌的生长。IL-22信号传导和岩藻糖基化的中断导致更易感染肠道和结肠炎,部分原因是条件致病菌的过度生长。
免疫系统常见抗体—IgA(有益菌的好伙伴)
IgA也能促进肠道细菌从上皮表面的分离,IgA是由肠道相关淋巴组织中的浆细胞产生。
大量的聚合物IgA在上皮细胞中跨通道进入内腔,其中IgA通过包被细菌和结合微生物抗原及其毒素来维持屏障功能,并塑造微生物的组成。
IgA反应在体内平衡过程中通过T细胞独立和T细胞依赖过程发生。IgA抗体具有典型的多反应性,与微生物脂多糖、DNA和鞭毛抗原结合亲和力低。通过Toll样受体(TLR)参与的微生物感测可以直接刺激IgA的产生,从而提供针对肠道炎症的保护。
【名词小讲堂】
Toll样受体(TLR):
· TLR是参与非特异性免疫(天然免疫)的一类重要蛋白质分子,也是连接非特异性免疫和特异性免疫的桥梁。
· TLR是单个的跨膜非催化性蛋白质,可以识别来源于微生物的具有保守结构的分子。
· 当微生物突破机体的物理屏障,如皮肤、粘膜等时,TLR可以识别它们并激活机体产生免疫细胞应答。
此外,T细胞对于MYD88对微生物信号的内在感应对体内IgA稳态反应对预防营养不良和肠病非常重要。
【名词小讲堂】
髓样分化因子(MYD88):是Toll样受体(TLR)信号通路中的一个关键接头分子,在传递上游信息和疾病发生发展中具有重要的作用。
肠道微生物群中的某些菌在结肠炎期间被IgA包被,当转移到无菌动物时,可增强对结肠炎的易感性,这表明在失调期间优先结合IgA可识别与IBD相关的菌群。
而大多数共生体强烈诱导T细胞独立的IgA结合,“非典型”细菌,如SFB,M. schaedleri,Prevotella spp.和Helicobacter sp. flexispira,可以逃避T细胞独立抗体应答,使其非常接近上皮表面,在上皮表面引发抗原依赖性,高亲和性,T细胞依赖性IgA反应。
这些研究表明,IgA可以通过促进有益共生菌的定殖来强化“健康”的微生物群落,但是,一旦在失调的状态下,可能会诱发IgA对潜在致病菌的免疫反应。
防止在粘膜中积聚的保护机制
前面小节提到的:促进共生体与上皮分离的保护机制并不是万无一失的。
考虑到存在于上皮表面附近的大量微生物,并不是所有的微生物都能守规矩,其中一小部分就可以在稳态条件下突破上皮屏障。为了应对这种情况,宿主免疫系统就发展出具有限制粘膜破坏性炎症和将渗透性微生物传播至全身组织的策略。
它可以让大量存在于上皮下面的特殊固有层巨噬细胞去杀死细菌共生体,通过多种机制吞噬和杀死渗透微生物,包括产生抗菌分子和活性氧。
在稳态条件下,这些驻留巨噬细胞在微生物感应方面有缺陷,因此不会引起炎症反应,这是一种通过IL-10刺激介导的调节活动。
肠道微生物也可以被固有层中的树突状细胞(DCs)吞噬并运输到肠系膜淋巴结,在那里,含有树突状细胞的细菌诱导保护性IgA和调节性T(Treg)细胞;这些树突状细胞不能到达全身的二级淋巴结结构,从而限制了它们的全身传播。
【名词小讲堂】
树状突细胞(DCs):
· 也称DC细胞,最早是由加拿大学者Steinman于1973年发现的
· 是功能最强的抗原提呈细胞,因其成熟时伸出许多树突样或伪足样突起而得名。
全身部位的控制
尽管前面强调存在坚固的粘膜防火墙,但依然会有非常小部分的肠道微生物可通过静脉门系统或血管系统扩散(图1)。 不过这些微生物到了那之后,肝脏会派出kupffer细胞或脾脏派出的巨噬细胞来把这些偷跑出来的菌吞噬和杀死。
共生菌还可以通过T细胞依赖和T细胞独立机制诱导IgG稳态反应。IgG抗体可以通过识别高度保守的蛋白质(例如murein脂蛋白)来结合多种细菌,包括变形杆菌和相关病原体,以限制细菌从肠道的全身传播。
此外,IgG2b和IgG3等型与IgA结合的共生物种具有相似的反应性,可以在母乳中转移给新生儿;这些母体IgG等型与IgA结合新生儿微生物群,以限制异常的共生特异性T细胞介导的炎症。
在缺乏IgA或MYD88和TIR结构域的动物中,系统性IgG对微生物群的反应增强,这些结构域包含用于TLR介导的细菌感应的干扰素-β(TRIF)信号适配器。
这种增强的IgG反应很可能代表由于适应性降低和渗透性共生细菌被杀死而导致的全身组织中共生菌增加的补偿性适应。
病原体的逃避
肠道病原体已经进化出多种策略来破坏和逃避粘膜防火墙,限制细菌共生体进入粘膜组织。几种病原体,包括肠沙门氏菌、Shigellaflexneri、Yersinia enterocolitica和霍乱弧菌,都会产生粘蛋白降解酶。
此外,肠道病原体已经进化出抵抗机制对抗IECs产生的抗菌蛋白。例如,肠杆菌可表达参与调节抗菌肽抗性的脂多糖修饰的基因,以及参与抗菌肽固存、外排和降解的基因。
同样,Listeria monocytogenes 使肽聚糖中的N-乙酰氨基葡糖残基脱乙酰化,从而逃脱了溶菌酶的溶菌活性,溶菌酶是Paneth细胞产生的一种酶。
与细菌共生体不同,肠道产生毒力蛋白,以逃避吞噬细胞中的溶酶体降解。进入肠细胞后,S. enterica 会破坏液泡膜并逃逸到宿主胞浆中,从而逃避自噬介导的降解,复制并在IECs之间传播。
在中性粒细胞中,S.flexneri 可以通过超氧化物歧化酶和过氧化氢酶的表达来抑制活性氧介导的杀伤。因此,肠道病原体已经进化出多种机制来逃避粘膜防火墙。也许与IBD更相关的是,在缺乏粘膜防火墙的一个或多个组件的易感宿主中,致病菌就有可能引发疾病。
IBD粘膜防火墙破裂
在上一小节,主要解释了坚固的粘膜防火墙,它是如何来保护宿主,在这一小节我们来看,在IBD患者中这个粘膜防火墙是如何被摧毁的。
减少微生物与上皮细胞表面接触的稳态过程的破坏可能会增加IBD发病的易感性,因为观察到多个IBD易感基因编码蛋白质,它们可以限制细菌共生体进入粘膜或促进细菌杀灭。
图2 炎症性肠病粘膜防火墙的破裂

炎症性肠病易感基因的突变会损害宿主用来防止有害微生物进入固有肠板的粘膜策略。
在小肠中,核苷酸结合寡聚结构域2(NOD2)和自噬相关基因(ATG16L1, IRGM, XBP1 and LRRK2)的变异导致Paneth细胞衍生抗菌肽(AMPS)的分泌减少。
肿瘤坏死因子15和IL23R基因多态性可影响第3组固有淋巴样细胞(ILC3s)和T辅助17(Th17)细胞,通过产生IL-22和IL-17从粘膜分离细菌共生体。
克罗恩病相关的吞噬细胞(如巨噬细胞和树突状细胞(DCs)中的NOD2功能丧失突变损害细菌识别。
肠上皮细胞自噬相关基因(ATG16L1, IRGM and NOD2)的突变可能导致细菌清除缺陷。
在大肠中,吞噬体烟酰胺腺嘌呤磷酸二核苷酸氧化酶复合物(CYBB, CYBA, NCF1, NCF2, NCF4, RAC1, RAC2)组分或调节因子的遗传变异损害了活性氧的氧化爆发和产生,导致吞噬细胞中细菌的杀伤缺陷。
功能丧失突变在IL10R改变肠道免疫稳态,导致失调的T细胞反应。
FUT2,岩藻糖基转移酶2;M,微凝胶;MUC2,粘蛋白2;SIgA,分泌性IgA;TGFβ,转化生长因子β;Treg细胞,调节性T细胞。
从上皮表面分离共生菌
在小鼠中,MUC2(图2左上角)的缺乏会导致粘液层异常,从而促进微生物群与肠上皮表面的紧密接近并导致自发性结肠炎。
在溃疡性结肠炎患者中观察到的共同特征是隐窝深处细菌与上皮紧密接触。
岩藻糖基转移酶2(FUT2)中的功能缺失变异,编码一种促进粘膜屏障功能的蛋白质,与克罗恩病易感性增加有关。
NOD2
核苷酸结合寡聚结构域2(NOD2)的遗传变异是第一个与克罗恩病相关的基因变异,也是已知的疾病发展的最强遗传危险因素。但光靠NOD2是不会感染的,它需要辅助条件。
NOD2是一种细胞内受体,能感受肽聚糖衍生的壁酰二肽并诱导对细菌的免疫反应。NOD2在Paneth细胞中表达,可能调节其抗菌功能。然而,Paneth细胞消融或基质金属蛋白酶7(MMP7)缺乏,可将不活跃的前α-防御素转化为杀菌形式,不会导致小鼠自发性炎症。
这表明IBD的发病机制需要额外的遗传缺陷或存在特定的致病菌,而这些致病菌在绝大多数在特定无病原体(SPF)条件下饲养的小鼠微生物群中均未发现。
克罗恩病与基因变异
克罗恩病相关蛋白自噬相关蛋白ATG16L1中的突变也可能通过损害Paneth细胞内分泌颗粒的胞吐作用而导致回肠疾病,这种活动限制了细菌共生菌的渗透。
未折叠蛋白反应(UPR)转录因子X-box结合蛋白1(XBP1)的基因变异与克罗恩病风险增加有关。XBP1特异性上皮缺失导致Paneth细胞内质网应激和结构缺陷。
值得注意的是,IECs中UPR和自噬通路的损伤与自发性克罗恩病(如跨壁回肠炎)有关。
两个克罗恩病易感基因,TNFSF15和IL23R,调节ILC3s和TH17细胞,它们在通过产生IL-17和IL-22抑制共生微生物方面起着至关重要的作用。
然而,还需要进一步的研究来了解这些基因的变异是如何与克罗恩病联系在一起的。
防止粘膜共生积累
利用固有层中的特殊大噬菌体杀灭细菌共生体,在限制黏膜损伤性炎症和防止渗透性微生物传播方面具有关键作用。
在肠道中,NOD2由吞噬细胞、上皮细胞、基质细胞和Paneth细胞表达。
值得注意的是,NOD2缺乏与CYBB(也被称为NOX2)的缺乏——吞噬体烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶复合物的一个组成部分,其通过氧化猝发杀死细菌-在小鼠中触发M. schaedleri的增殖和自发性克罗恩病样TH1细胞驱动性结肠炎。
由于克罗恩病相关的NOD2变异体是功能丧失突变,因此吞噬细胞或肠和/或基质细胞内产生的细菌感应减弱可能促进病理生物的管腔累积和粘膜渗透,导致T细胞介导的肠道炎症。
然而,NOD2相关的克罗恩病是否是由人类体内特定病理生物的积累引起的,还需要进一步的研究。
NOD2在自噬中也有作用,自噬是一种介导溶酶体降解和细胞内细菌清除的途径。
NOD2将克罗恩病相关蛋白ATG16L1募集到细菌进入部位的质膜上,但克罗恩病相关的NOD2变异体有缺陷的ATG16L1募集和受损的细菌诱导的上皮内自噬。
除ATG16L1外,免疫相关GTPase家族M(IRGM)和富含亮氨酸重复激酶2(LRRK2)也调节自噬途径,这些基因的变化与克罗恩病风险有关。
由于自噬的缺陷损害了细胞内细菌的清除能力,可以想象,自噬相关基因的突变可能导致病理生物在肠道中的渗透和炎症。
在极早发性IBD患者中,细菌感染与IBD易感性之间的联系是一个明显的例子,它与吞噬细胞NADPH氧化酶复合物的组分和调节基因的遗传变异有关。
同样,高达40%的慢性肉芽肿性疾病,由NADPH氧化酶组分的功能突变导致的原发性免疫缺陷病,发展为克罗恩病样结肠炎。
总的来说,这些观察表明,杀死细菌共生体的缺陷促进了克罗恩病的发展。
共生的有益影响
共生细菌通过增强粘膜防火墙来促进保护性免疫,这些防火墙限制了共生体的渗透,同时控制了对微生物的异常T细胞反应。破坏这些相互作用的宿主-共生相互作用可能会促进对IBD患者的失调菌群的不适当免疫反应。
诱导保护性免疫反应
肠道微生物群在调节宿主免疫以建立和维持肠道稳态方面起着至关重要的作用。
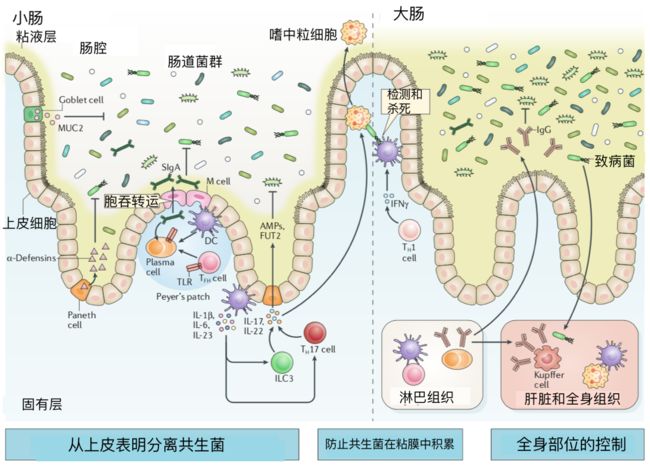
图3 共生体的有益作用。
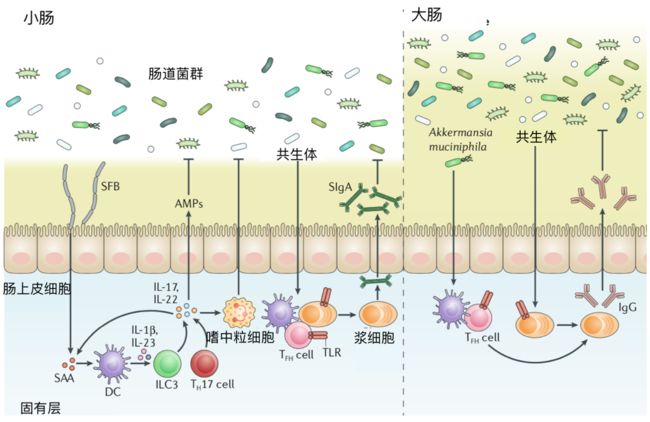
分段丝状细菌(SFB)与肠上皮的粘附会触发血清淀粉样蛋白A(SAA)的释放,后者作用于固有层的树突状细胞(DCs)上,以刺激包括IL-1β和IL-23在内的细胞因子的分泌,并诱导视黄酸受体相关的孤儿受体-γt (RORγt)+ T辅助物17(TH17)细胞分化和第3组先天性淋巴样细胞(ILC3)激活。
由ILC3S分泌的IL-22增强了上皮的SAA产生,以增强Th17细胞介导的粘膜防御,包括抗菌肽(AMP)分泌和中性粒细胞募集。
共生体还通过Toll样受体(TLRs)诱导的B细胞内和T细胞内微生物传感诱导的IgA反应促进肠道稳态。分泌的IgA(SIGA)通过与肠道微生物结合和防止上皮易位而促进屏障功能。
同样,IgG的产生可以通过TLR对B细胞的参与直接发生,而粘液阿克曼菌Akkermansia muciniphila的肠道定植可以通过抗原特异性T滤泡辅助(TFH)细胞反应诱导稳态IgG的产生。此外,IgG有助于穿透上皮屏障的病原体的全身控制(未显示)。
限制不适当的炎症
Treg细胞起源于两种不同的个体遗传谱系:
胸腺来源的Treg(tTreg)细胞
外周来源的Treg(pTreg)细胞
大多数pTreg细胞在共生菌的存在下在胸腺外的结肠中发育,因为在无菌小鼠中,结肠中pTreg细胞的频率明显降低。
Treg细胞通过防止诱导不适当地的T细胞对微生物抗原的反应而在维持组织稳态方面起着至关重要的作用(图4a)
动物:
例如小鼠,缺乏对其调节或功能有重要作用的Treg细胞或因子(包括IL-10、转化生长因子β(TGFβ)和αVβ8整合素)的小鼠会发生自发性结肠炎。
人类:
与动物实验数据一致,IL-10受体突变的幼童会发展成结肠克罗恩病。
此外,FOXP3是Treg细胞发育所需的转录因子,其突变的小鼠和人类都有自身免疫性疾病,包括结肠炎。
图4 调节性T细胞支持肠道稳态
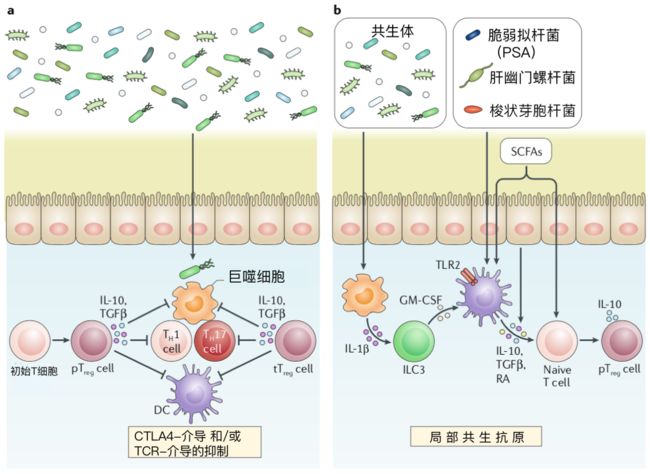
a | 外周来源的调节性T(pTreg)细胞和胸腺来源的Treg(tTreg)细胞抑制肠道异常炎症。Treg细胞产生IL-10和转化生长因子β(TGFβ),以抑制效应T辅助1(TH1)细胞和TH17细胞。另外,髓样细胞如巨噬细胞是IL-10驱动的肠内稳态的重要靶标。
肠道Treg细胞还通过抑制通过细胞毒性T淋巴细胞抗原4(CTLA4)介导的或T细胞受体(TCR)介导的抗原呈递细胞(例如树突状细胞(DCs))来抑制效应T细胞反应。自反应性tTreg细胞也可通过与微生物抗原的TCR交叉反应来促进抗原特异性免疫抑制。
b |共生体通过抗原依赖性和非抗原依赖性过程在pTreg细胞的外周培养中起着至关重要的作用。
细菌的巨噬细胞感应导致IL1β刺激的第3组先天淋巴样细胞(ILC3s)分泌粒细胞-巨噬细胞集落刺激因子(GMCSF),进而增强视黄酸(RA)和IL10的DCs表达,以支持pTreg细胞分化和在结肠中扩散。
此外,除了RA和IL10外,共生体还可以通过上皮细胞和DC促进TGFβ的表达。脆弱拟杆菌可通过与Treg细胞和DC表达的Toll样受体2(TLR2)结合的多糖A(PSA)增强Treg细胞的活性。
选择细菌种类可以直接诱导pTreg细胞分化;这些pTreg细胞对梭状芽胞杆菌和螺杆菌表达的抗原具有TCR特异性。这表明共生抗原在支持Treg细胞分化中起作用。
短链脂肪酸(SCFAs)是包括梭状芽孢杆菌在内的共生菌产生的发酵副产物,可通过表观遗传修饰或通过增强RA产生的DCs直接促进pTreg细胞群的扩张。
Treg细胞在抑制共生菌免疫反应中的作用也得到了证实,通过Treg细胞的共同转移,CD45RBhiCD4+T细胞转移到淋巴细胞减少小鼠中而引起的微生物依赖性结肠炎被清除。
pTreg细胞缺乏动物粘膜屏障的免疫病理学表明,tTreg细胞足以系统地维持对自身抗原的耐受性,而pTreg细胞在抑制肠道炎症方面具有非冗余的作用。
结肠pTreg细胞深受局部抗原的影响,TCR repertoire 不同于外周淋巴结和脾脏的类似Treg细胞,能够识别梭状芽孢杆菌和拟杆菌属表达的抗原,这进一步支持细菌对肠Treg细胞诱导的重要性(图4b)
IBD中共生关系的破坏
大量的证据表明,微生物群在引发IBD方面起着至关重要的作用。此外,与IBD相关的遗传缺陷与环境因素一起,可以诱导病理离子的积累和渗透到肠道,从而进一步促进肠道菌群失调和炎症反应。
引起IBD的微生物群
尽管没有一种病原体或致病菌被一致认为是IBD的病因,但多方面的证据支持微生物群在驱动肠道炎症中的重要作用。例如,粪便流改道可以减少或消除了回肠克罗恩氏病的炎症。
此外,在回肠切除术后,克罗恩病的复发取决于暴露于肠腔内容物。抗生素治疗可使活动期溃疡性结肠炎和克罗恩病患者病情缓解,并可预防某些克罗恩病患者复发。然而,考虑到这些药物对多种菌群的影响,抗生素研究的解释是困难的。
动物模型进一步支持肠道微生物群在IBD发病机制中的作用。例如,将结肠炎小鼠的粪便微生物群口服到健康动物体内就足以引发疾病。最重要的是,基因易感的小鼠在常规的微生物群的情况患上了结肠炎,但不是在无菌条件下。例如,SPF小鼠,而不是无菌小鼠,TCR基因缺失突变会导致结肠炎。
此外,在无菌的TCR缺陷小鼠中没有观察到结肠炎,这些小鼠被固定在一个确定的共栖群落中,这表明引发疾病需要特定的微生物。
IBD患者的菌群失调
对IBD患者体内微生物群的组成进行了广泛的研究。16SrRNA测序分析粪便和粘膜样本显示存在菌群失调,其特征是菌群多样性下降,厚壁菌门的某些属的改变和肠杆菌科物种的丰度增加。
这些改变在克罗恩病患者中比溃疡性结肠炎患者更为明显。一些研究还显示了拟杆菌属的变化,特别是在克罗恩病患者中。
研究发现,IBD患者与近亲(包括双胞胎)之间的微生物群也存在差异,这表明菌群失调与疾病有关,而不是与遗传因素有关。
然而,需要纵向研究来确定是否失调先于炎症的发生。与未治疗的克罗恩病患者在诊断时收集的粪便样本相比,粘膜样本中微生物群的失调更为明显。与溃疡性结肠炎相比,对粘膜样本的研究还显示克罗恩病中潜在有益菌的净损失更大,某些菌群包括克罗恩病中的肠杆菌科和溃疡性结肠炎中的瘤胃球菌属与疾病活动性的相关,以及克罗恩病中某些菌群与肿瘤坏死因子治疗反应的相关。
粪便样本的宏基因组测序显示,克罗恩病患者和非IBD患者之间存在明显的区分,而溃疡性结肠炎患者的差异更显著,总体上有菌群多样性丧失的趋势。
这些研究还发现IBD患者粪便中代谢物多样性的丧失与菌群多样性的丧失相当。尽管到目前为止,元基因组学的研究仅限于粪便样本的分析,但由于宿主来源的DNA在粘膜活检样本中含量很高,便于更深入理解IBD中微生物群的功能破坏。
其他微生物——真菌
虽然IBD中的大多数微生物组学研究都集中在细菌对疾病发病的贡献上,但也有一些研究强调了其他微生物在IBD中的重要性。例如,与健康人相比,克罗恩病患者结肠活检样本中真菌菌群多样性增加。同样,克罗恩病患者回肠粘膜标本和粪便标本中的真菌多样性增加,白念珠菌、棒状曲霉菌和新生隐球菌增加。
其他证据证实了IBD中真菌失调的存在,包括与健康受试者相比,酿酒酵母菌比例降低,白色念珠菌增加。值得注意的是,一些研究将真菌菌群多样性的增加与疾病的严重程度联系起来,并认为克罗恩病的环境有利于真菌而损害了细菌,或者抗生素治疗为真菌扩张创造了特定的生态位。
肠道真菌与宿主免疫受体dectin 1相互作用,该信号通过CARD9诱导炎症分子的产生和TH17细胞的反应。CARD9变异与发生IBD的风险增加有关。同样,Card9基因敲除小鼠改变了肠道真菌群落,增加了化学诱导结肠炎的易感性。此外,dectin 1基因的多态性与医学上难治的溃疡性结肠炎有关。
其他微生物——病毒
肠道中也含有大量的病毒,这些病毒可能在IBD的发病机制中起作用。例如,感染小鼠诺如病毒会导致潘氏细胞异常,出现克罗恩病易感基因ATG16L1的突变。
此外,FUT2的遗传变异与诺如病毒感染和克罗恩氏病的易感性有关。 可能与IBD相关的其他病毒是噬菌体。
IBD患者肠道病毒组的宏基因组测序显示,与对照组相比,Caudovirales 噬菌体有表达,这似乎不是继发于细菌菌群的变化;然而,这种扩增是队列特异性的,未在验证队列中得到证实。因此,需要更多的研究来了解真菌和病毒在人类IBD中的作用。
IBD 的前因后果
虽然在IBD早期微生物群的改变可以独立于治疗而发生,但没有直接证据表明生态失调在IBD发病机制中的因果作用。
在小鼠中,化学诱导的结肠炎和肠道感染会引发微生物群组成的强烈变化,其中一些变化与在IBD患者中观察到的变化相当,包括变形菌的增殖。
炎症导致肠腔氧合增加,硝酸盐和宿主电子受体的有效性增加,可驱动肠杆菌科的厌氧呼吸和增殖。
因此,在IBD患者的肠粘膜和肠腔中观察到的许多微生物群变化可能是继发于炎症。
在一个自发性克罗恩病的遗传模型中,纵向分析揭示了在结肠炎发病前单一或有限数量的致病菌的种群扩张,这种菌群足以在具有复杂微生物群的小鼠中引发疾病。
根据这些观察,失调可能分为早期和晚期两个阶段进行。
在早期,IBD相关的遗传和环境因素可能导致致病菌的积累,这可能先于临床疾病的发展。
尽管参与早期失调的致病菌的身份和数量尚不清楚,但在IBD动物模型中有限的证据表明,该细菌的遗传和代谢特征可能是重要的。
例如,schaedleri和Helicobacter hepaticus,这两种致病菌可以在遗传易感性小鼠中引发自发性结肠炎,产生毒力因子并生活在上皮附近,尽管这些活性在诱发结肠炎中的作用尚不清楚。当黏膜防火墙受到IBD易感基因突变的影响时,这种细菌特征可能降低局部穿透的阈值(图5)
图5 炎症性肠病中的生态失调

与炎症性肠病相关的遗传缺陷,再加上饮食和抗生素使用等环境因素,可能导致引起疾病的病原体积累和渗透到肠道固有层(早期失调)中,这可能先于临床显性疾病的发展。
炎症可导致细菌类群的更大变化,包括变形菌(晚期失调)的扩张,其途径是增强肠腔氧合,增加炎症肠道环境中的硝酸盐(NO3-)、宿主来源的氧受体和铁的可用性。
这种后期的失调还表现为微生物多样性全面下降,有益的共生体丧失,这可能导致粘膜粘附增加,共生微生物移位,从而引发慢性炎症。
DC,树突状细胞;TH细胞,T辅助细胞;Treg细胞,调节性T细胞;SCFAs,短链脂肪酸。
在晚期失调期间,肠道炎症推动了菌群的进一步变化,包括变形菌的增殖(图5)。鉴于肠道微生物群的不同菌群对宿主免疫系统和肠道屏障有有益的影响,某些菌群的缺失可能导致肠道炎症的加剧或消退。特定细菌的大量繁殖,如粘附性和侵袭性大肠杆菌,积聚在IBD患者的炎症粘膜中,可进一步促进炎症反应(图5)。
粘附性和侵入性大肠杆菌使用常见的1型菌毛粘附素FimH粘附到肠上皮,该粘附素可识别在克罗恩病患者回肠中异常表达的癌胚抗原相关细胞粘附分子6(CEACAM6)。 需要对来自IBD易感性增高的人群和/或IBD患者的黏膜样本进行精心设计的纵向研究,以了解其在疾病复发中的作用。
IBD患者对微生物抗原的适应性免疫应答
IBD患者常检测到T细胞和抗体对微量双抗原的反应增强,进一步证明了微生物群在疾病发病机制中的作用。与健康人相比,IBD患者产生大量抗共生细菌的IgG抗体,并且血清中抗真菌和/或细菌肽和聚糖的活性较高。尽管抗微生物抗体可以作为疾病和诊断的标志物,但这种抗体在IBD中的作用尚不清楚。鉴于健康的小鼠和人类产生了抗细菌共生体的抗体,IBD相关抗体高滴度的存在可能反映了IBD患者的适应性免疫反应增强或肠道内微生物抗原暴露增加。
除了体液反应外,在IBD患者中还观察到T细胞对微生物群的反应失调。早期研究表明,IBD患者炎症粘膜中的T细胞或单核细胞在肠道微生物抗原刺激后,其反应性较正常粘膜中的细胞有所增强。
然而,在IBD患者和健康人的血液和粘膜中都可以检测到CD4+T细胞对细菌共生体的反应。对微生物群有反应的人T细胞主要具有记忆表型,具有不同的TCR Vβ基因库,这与对多种微生物抗原的反应一致。
与人类数据一致的是,在结肠炎动物模型中也可以检测到对共生细菌和细菌抗原有反应的T细胞。CD4+T细胞对共有抗原的反应转移到淋巴细胞减少的动物体内足以引发结肠炎。
一些实验观察结果表明,致病性CD4+TH细胞可以在稳态条件下识别致病生物,而Treg细胞介导的免疫抑制对结肠炎的发展至关重要。然而,Treg细胞作用于效应T细胞抑制细菌引起的肠道炎症的机制尚不清楚。Treg细胞可以以抗原无关的方式抑制效应T细胞和/或通过其TCR作用于抗原提呈细胞以抑制抗原特异性效应T细胞。
针对微生物群进行治疗
目前大多数IBD的治疗方法,包括类固醇和生物药物,如抗肿瘤坏死因子或抗整合素治疗,都能抑制宿主免疫系统,但不直接针对引起或导致炎症的微生物。
鉴于炎症性肠病的治疗在不到50%的患者中可获得完全缓解,针对肠道微生物的治疗方法的发展可能为治疗炎症性肠病提供一种独特的方法。
图6 微生物治疗在炎症性肠病中的作用
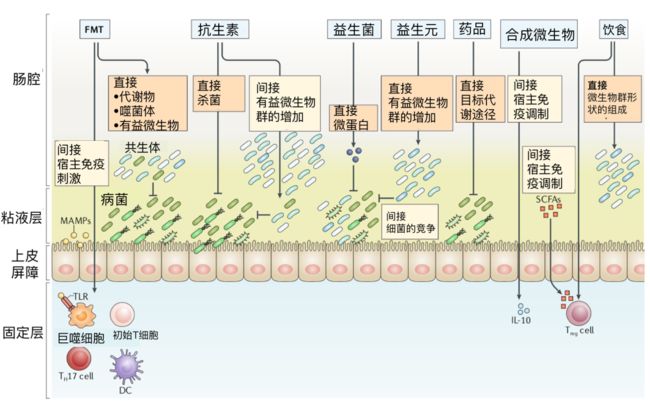
鉴于肠道微生物群在炎症性肠病发病机制中的关键作用,针对肠道微生物的治疗方法的发展可能为治疗该疾病提供一种独特的方法。
粪便菌群移植(FMT)
直接有益效果:通过转移细菌衍生的代谢产物和噬菌体或通过恢复有益微生物而产生直接的有益效果。
间接有益效果:通过细菌成分间接产生有益的效果。例如,Toll样受体(TLR)由微生物相关分子模式(MAMPs)触发宿主固有免疫刺激。
抗生素
抗生素可能通过直接杀死致病菌或间接促进有益微生物的种群扩张而产生广泛的作用。
益生菌
使用益生菌制剂,如大肠杆菌Nissle 1917,可导致微球蛋白的分泌,微球蛋白是具有抗菌活性的小多肽,可直接抑制与肠杆菌科密切相关的细菌。
益生元
补充益生元可以促进有益共生菌的种群扩张,从而战胜有害细菌。
其他
合成工程微生物的给药可以通过调节免疫系统(例如通过分泌IL-10)对宿主产生有益作用。
饮食通过促进有益微生物的生长或减少致病微生物的消耗来塑造肠道微生物群的组成。
来自膳食多糖的短链脂肪酸(SCFA)也可以支持肠道调节性T(Treg)细胞群的分化和扩展。
随着对某些微生物的失调和免疫调节功能的进一步了解,研究人员为改善肠道微生物群以预防或改善IBD提出了一些创新的策略。
粪便微生物群移植(FMT)
IBD和反复难辨梭状芽孢杆菌感染的生物学特性:
门级多样性的丧失和肠杆菌科兼性厌氧菌(包括变形菌)的过度生长。因此研究人员想到了纠正微生物群落失衡可能会成为治疗IBD的有效方法。
临床效果
通过恢复肠道微生物群落多样性,粪便微生物群移植(FMT)在解决复发性艰难梭菌感染方面的高成功率增强了微生物群调节疗法的潜力。尽管已经接受FMT治疗的IBD患者数量有限并且对治疗的反应有所不同,但大约30%的溃疡性结肠炎患者已经实现了临床缓解。抗生素预处理和多次粪便输注似乎可以改善FMT在溃疡性结肠炎患者中的有效性。
也有研究人员对此持不同看法。
Rossen等人的一项双盲,随机,安慰剂对照研究未观察到FMT对溃疡性结肠炎患者有任何有益作用,其结果发表在Gastroenterology期刊。
此外,FMT在克罗恩病患者中的有效性仍不清楚,可能是因为目前的研究报告潜在疗效在队列规模上是有限的,并且缺乏安慰剂治疗组。需要进一步的研究,了解更多机制,来制定更有效的治疗方案。
抗生素
随机对照试验的Meta分析显示了抗生素在治疗克罗恩病和溃疡性结肠炎中的益处。
然而,使用抗生素来改变微生物群受到限制,因为在消灭致病菌的同时对有益菌也有影响,这可能带来不良后果。
由于相互矛盾的发现,在IBD治疗中抗生素的使用仍然有限,但使用广谱抗生素混合疗法的研究表明,它们可能重症急性结肠炎和慢性溃疡性结肠炎有效。
在未来,一旦确认了引起IBD的微生物代谢途径,就有可能用新一代抗生素专门针对它们。

益生菌
活微生物可能在几个方面是有利的。
一旦达到稳定的定殖,有益的微生物因子可以持续地传递给宿主,活菌可能会触发有益的免疫反应。通过服用益生菌,包括乳酸菌或双歧杆菌,有针对性地调节微生物群,已经成功地治疗了一些肠道疾病。
此外,在维持溃疡性结肠炎患者病情缓解方面,益生菌大肠杆菌Nissle 1917口服治疗与美沙拉嗪标准治疗具有相似的疗效。益生菌大肠杆菌Nissle 1917能分泌具有抗菌活性的微胞素,抑制可能加剧肠道炎症的竞争性肠杆菌科细菌。

但是,益生菌通常对微生物组的整体组成影响有限,因为这些细菌无法持久地定居健康的成年人。 在某些适应症中,同时补充益生元可以提高外源微生物的移入效率,包括增强对艰难梭菌的定植性。
合成生物学的进展为IBD的治疗和管理提供了创新的方法(如下图6)
给小鼠口服经工程改造以表达和分泌IL-10的乳酸乳球菌,足以保护它免受DSS和IL-10缺乏引起的结肠炎的侵害。
在一小群克罗恩氏病患者中,使用限制胸腺嘧啶的遏制策略安全地生产了重组的产生IL-10的乳酸乳球菌菌株,但仅引起了很小的疾病活动性改善。
由于乳酸杆菌的惰性,这种益生菌也被修饰以表达胰岛素生长因子1、血红素氧合酶1或丝氨酸蛋白酶抑制剂;当给小鼠口服时,这些合成的益生菌可以缓解实验性结肠炎的发展。然而,这些策略在临床试验中并不成功,可能是由于工程乳酸菌菌株不能持续地将IBD患者定殖。
靶向微生物代谢
精确定位有害微生物在失调期间增殖的代谢途径,对于改善小鼠结肠炎是有效的。例如,变形杆菌利用增加的氮源,包括在炎症肠中产生的一氧化氮,大量繁殖。
值得注意的是,靶向钼辅因子依赖的酶,需要使用一氧化氮进行厌氧呼吸,通过减弱肠杆菌科的增殖,包括变形菌,来保护小鼠免受DSS诱导的结肠炎。因此,开发针对有害菌代谢途径的药物可能为治疗IBD提供一种新的策略。
饮食
饮食在塑造微生物群的组成中起着重要作用,并可通过调节饮食来控制IBD症状。
全肠内营养(EEN)是IBD中为数不多的已被广泛研究的膳食干预措施之一,它是一种营养完整的无固体食物的元素和聚合物配方食品。在克罗恩病的儿科患者中,甚至与皮质类固醇一样有效,但没有与皮质类固醇治疗相关的副作用。

EEN能独立于其他环境因素迅速改变微生物群的组成,有效降低克罗恩病患儿的肠道炎症。EEN诱导克罗恩病缓解的机制尚不清楚,但它可能促进有益微生物的生长或减少致病生物。
在实验性DSS诱导的结肠炎中,一项对各种精制饮食的调查确定了洋车前子纤维的有益作用。而膳食蛋白(包括酪蛋白)的增加,粪便微生物密度的增加以及肠通透性的改变加剧了结肠炎的严重性。
寄主对结肠炎的易感性很大程度上依赖于形成微生物群的特定纤维或蛋白质成分的组合,可见饮食成分在IBD中非常重要。
结 语
在过去的十年里,人们关于微生物群-宿主相互作用以及遗传和免疫系统在IBD中的作用的了解有了大幅提高。然而,对于IBD发病机制的几个方面的认识仍很局限。
一个主要的问题是缺乏对引起易感个体炎症的IBD致病菌的识别。多种致病菌可能引起疾病,这些致病菌可能因患者特定的IBD易感位点而异。因此,利用具有相同或相似遗传缺陷的个体进行研究可能对鉴定这种微生物很重要。
识别引起IBD的微生物对于理解疾病的发病机制,监测患者这些病原体的改变以及合理开发新疗法具有重要意义。
动物模型为研究微生物群和免疫系统在疾病发展中的作用提供了重要的线索。然而,除了少数之外,这些模型并不是基于与人类疾病相关的遗传缺陷,也没有完全概括IBD的病理学。小鼠模型通常使用简化的微生物群,这种简化的微生物群不能反映完整肠道中发生的复杂的微生物-宿主相互作用。
未来,如果能开发出更适合人类IBD的新模型,掌握更多机制,或许能从微生物群的角度(比如说饮食干预)带来更多的治疗甚至是预防措施。
参 考 文 献
Caruso R, Lo B C, Núñez G. Host–microbiota interactions in inflammatory bowel disease[J]. Nature Reviews Immunology, 2020: 1-16.
欢迎关注:谷禾健康
——让你和你的家人更健康