1.比对
###sample:GK10-HY
gzip -dc /Duhongli/Harddisk1/data/HY-3-4wk/GK10-HY/GK10-HY_R1.fq.gz >/Duhongli/Harddisk2/cuiying/HY/GK10-HY_R1.fq
gzip -dc /Duhongli/Harddisk1/data/HY-3-4wk/GK10-HY/GK10-HY_R2.fq.gz >/Duhongli/Harddisk2/cuiying/HY/GK10-HY_R2.fq
mkdir HY_GK10
STAR --runThreadN 16 --genomeDir /Duhongli/Harddisk1/Rat6.0genome/ensembl/STAR-index/GenomeDir --readFilesIn GK10-HY_R1.fq GK10-HY_R2.fq --quantMode GeneCounts --outSAMstrandField intronMotif --outSAMtype BAM SortedByCoordinate --outFileNamePrefix ./HY_GK10 --twopassMode Basic
rm -rf GK10-HY_R1.fq GK10-HY_R2.fq
结果文件:
HY_GK10Log.final.out:比对结束后的比对统计的信息

HY_GK10Log.out:记录了程序运行时的信息,可以用来回溯错误
HY_GK10Log.progress.out:报告对比进程信息,每分钟更新一次
HY_GK10ReadsPerGene.out.tab

HY_GK10SJ.out.tab:包含剪切信息

col1:chromosome
col2:first base of the intron
col3:last base of the intron
col4:strand(0:undened;1:+;2:-)
col5:intron motif------0:non-canonical;1:GT/AG;2:CT/AC;3:GC/AG;4:CT/GC;5:AT/AC;6:GT/AT
col6:0:unanotated;1:annotated(only if splice junctions database is used)
col7:num of the uniquely mapping reads crossing the junction
col8:num of the multi-mapping reads crossing the junction
col9:maximum spliced alignment overhang
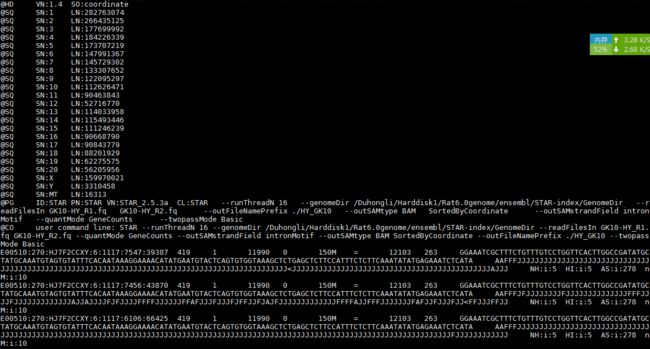
HY_GK10Aligned.sortedByCoord.out.bam
samtools view -h HY_GK10Aligned.sortedByCoord.out.bam
2、组装
###sample:HYGK10
stringtie /Duhongli/Harddisk2/cuiying/HY/HY_GK10Aligned.sortedByCoord.out.bam -b HY_GK10 -e -G /Duhongli/Harddisk1/Rat6.0genome/ensembl/STAR-index/Rattus_norvegicus.Rnor_6.0.86.chr.gtf -p 10 -o gtf/HY_GK10.stringtie.gtf
结果文件
-o HY_GK10.stringtie.gtf

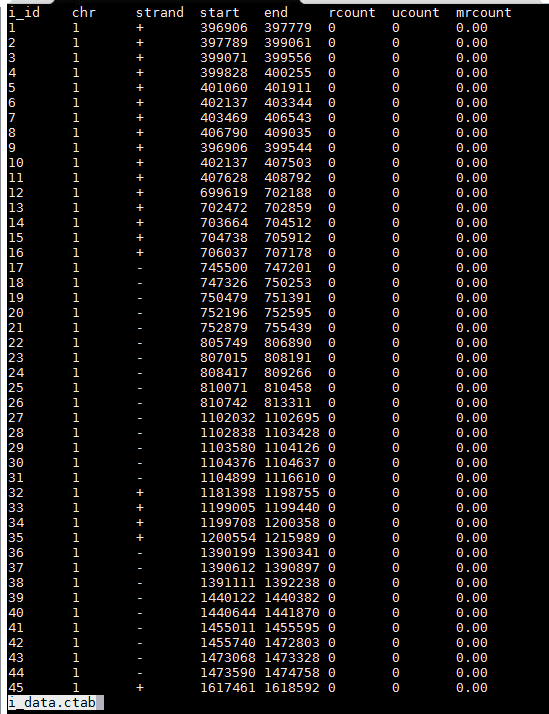
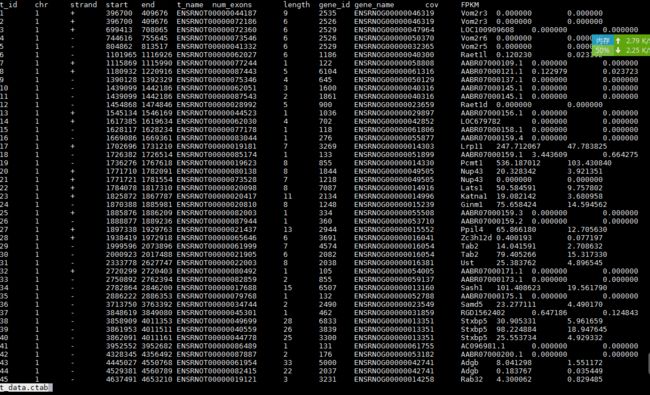
-b e2t.ctab;e_data.ctab;i2t.ctab;i_data.ctab;t_data.ctab

3、差异表达分析
source("http://bioconductor.org/biocLite.R")
biocLite("ballgown")
library(ballgown)
data_directory="/Duhongli/Harddisk2/cuiying/HY/stringtie_result/";
bg<-ballgown(dataDir=data_directory,samplePattern="HY",meas="all")
transcript_fpkm=texpr(bg,'FPKM')
gene_expression = gexpr(bg)
whole_tx_table = texpr(bg, 'all')
source("/Duhongli/Harddisk1/Test2018/bayesreg.R")
T<-bayesT(gene_expression,numC=3, numE=3,ppde=TRUE,betaFit=1, bayes=TRUE, doMulttest=1)
write.table(gene_expression, file="gene_expression.txt",quote=F, sep="\t")
write.table(whole_tx_table, file="whole_tx_table.txt",quote=F, sep="\t")
write.table(T, file="gene_expression.ttest.txt",quote=F, sep="\t")
q()
perl /Duhongli/Harddisk1/Test2018/deal_bayesT.pl
gene_expression.ttest.txt |awk '{print $1}'>diff.gene.txt

4、vcf
#对bam文件加头
java -jar /Duhongli/bin/GATK-Picard/picard.jar AddOrReplaceReadGroups INPUT=/Duhongli/Harddisk2/cuiying/HY/HY_bam/HY_GK10Aligned.sortedByCoord.out.bam OUTPUT=HY_GK10.RG.bam RGID=hy_gk10 RGLB=hy.RNAseq RGPL=illumina RGPU=HISEQ RGSM=HY_GK10
#标记重复
java -jar /Duhongli/bin/GATK-Picard/picard.jar MarkDuplicates I=HY_GK10.RG.bam O=HY_GK10.RG.dp.bam CREATE_INDEX=true VALIDATION_STRINGENCY=SILENT M=HY_GK10.metrics
#去N
java -jar /Duhongli/bin/GATK-Picard/GenomeAnalysisTK.jar -T SplitNCigarReads -R /Duhongli/Harddisk1/Rat6.0genome/ensembl/STAR-index/rn6.fa -I HY_GK10.RG.dp.bam -o HY_GK10.RG.dp.spN.bam -rf ReassignOneMappingQuality -RMQF 255 -RMQT 60 -U ALLOW_N_CIGAR_READS
#variant calling
java -jar /Duhongli/bin/GATK-Picard/GenomeAnalysisTK.jar -T HaplotypeCaller -R /Duhongli/Harddisk1/Rat6.0genome/ensembl/STAR-index/rn6.fa -I HY_GK10.RG.dp.spN.bam -dontUseSoftClippedBases -stand_call_conf 20.0 -o HY_GK10.vcf
#variant filtering
java -jar /Duhongli/bin/GATK-Picard/GenomeAnalysisTK.jar -T VariantFiltration -R /Duhongli/Harddisk1/Rat6.0genome/ensembl/STAR-index/rn6.fa -V HY_GK10.vcf -window 35 -cluster 3 -filterName FS -filter "FS > 30.0" -filterName QD -filter "QD < 2.0" -o HY_GK10.fit.vcf
rm -rf HY_GK10.RG.bam HY_GK10.RG.dp.bam HY_GK10.vcf
