文章是:Identification of ankylosing spondylitis-associated genes by expression profiling ,数据集是GSE25101。
文章中没有给出logFC的阈值,但是最后得到的差异基因中74个上调,9个下调,所以我们可以自己设置阈值,是得到这样的基因数目,然后得到的差异基因再应用于后面富集分析中d

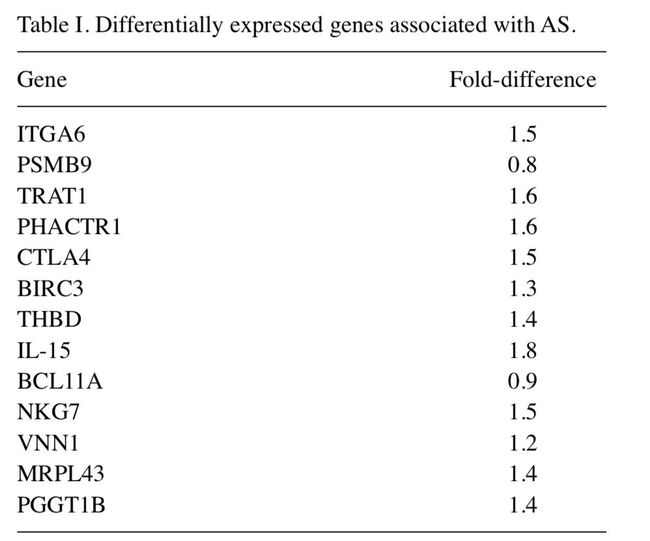
如上图所示,作者得到的13个在某些通路中(上图中黄色标注中间的部分)有富集到的基因如下图所示,所以我们需要做富集分析,得到有统计学意义的基因并和原文比较。
代码如下,其中下载和注释都是用的老大的新写的包哦,如下:
GEO数据库中国区镜像横空出世
第一个万能芯片探针ID注释平台R包
第二个万能芯片探针ID注释平台R包
第三个万能芯片探针ID注释平台R包
代码参考如下
library(devtools)
install_github("jmzeng1314/GEOmirror")
library(GEOmirror)
geoChina('GSE25101')
load('GSE25101_eSet.Rdata')
a=gset[[1]]
e=exprs(a)
p=pData(a)
gpl=a@annotation
gpl
#install_github("jmzeng1314/idmap1")
library(idmap1)
ids=getIDs(gpl)
group_list=c(rep('normal',16),rep('AS',16))
probe2gene=ids
probes_expr=e
filterEM <- function(probes_expr,probe2gene){
probe2gene <- probe2gene[probe2gene$probe_id %in% rownames(probes_expr),]
probes_expr <- probes_expr[match(probe2gene$probe_id,rownames(probes_expr)),]
}
genes_expr=filterEM(probes_expr,probe2gene)
boxplot(genes_expr)
library(limma)
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
head(design)
exprSet=genes_expr
rownames(design)=colnames(exprSet)
design
contrast.matrix<-makeContrasts("AS-normal",
levels = design)
contrast.matrix ##这个矩阵声明,我们要把 Tumor 组跟 Normal 进行差异分析比较
colnames(design)
deg = function(exprSet,design,contrast.matrix){
##step1
fit <- lmFit(exprSet,design)
##step2
fit2 <- contrasts.fit(fit, contrast.matrix)
##这一步很重要,大家可以自行看看效果
fit2 <- eBayes(fit2) ## default no trend !!!
##eBayes() with trend=TRUE
##step3
tempOutput = topTable(fit2, coef=1, n=Inf)
nrDEG = na.omit(tempOutput)
#write.csv(nrDEG2,"limma_notrend.results.csv",quote = F)
head(nrDEG)
return(nrDEG)
}
deg = deg(exprSet,design,contrast.matrix)
DEG <- deg
DEG$probe_id = rownames(DEG)
merge_DEG <- merge(DEG,probe2gene,by='probe_id')
save(merge_DEG,file = 'merge_DEG.Rdata')
## for volcano plot
df=merge_DEG
attach(df)
df$v= -log10(P.Value)
mean(abs(df$logFC))+2*sd(abs(df$logFC))
df$g=ifelse(df$P.Value>0.05,'stable',
ifelse( df$logFC >0.6,'up',
ifelse( df$logFC < -0.,'down','stable') )
)
table(df$g)
df$name=rownames(df)
head(df)
ggpubr::ggscatter(df, x = "logFC", y = "v", color = "g",size = 0.5,
label = "name", repel = T,
label.select =head(df$symbol),
palette = c("#00AFBB", "#E7B800", "#FC4E07") )
detach(df)
# x=merge_DEG$logFC
# names(x)=rownames(DEG)
# cg=c(names(head(sort(x),100)),
# names(tail(sort(x),100)))
# cg
# library(pheatmap)
# n=t(scale(t(genes_expr[cg,])))
# n[n>2]=2
# n[n< -2]= -2
# n[1:4,1:4]
# ac=data.frame(groupList=groupList)
# rownames(ac)=colnames(n)
# pheatmap(n,show_colnames =F,show_rownames = F,
# annotation_col=ac)
# DEG['ILMN_1757298',]
# DEG['ILMN_1681591',]
# boxplot(as.numeric(genes_expr['ILMN_1757298',])~groupList)
library(ggplot2)
library(clusterProfiler)
tmp_merge_DEG <- bitr(merge_DEG$symbol, fromType="SYMBOL",
toType="ENTREZID",
OrgDb="org.Hs.eg.db")
merge_DEG_new <- merge(merge_DEG,tmp_merge_DEG,by.x='symbol',by.y='SYMBOL')
mdn <- merge_DEG_new
mdn$change = ifelse(mdn$adj.P.Val < 0.05 & abs(mdn$logFC) > 0.5,
ifelse(mdn$logFC > 0.5 ,'UP','DOWN'),'NOT')
save(mdn,file = 'mdn.Rdata')
load('mdn.Rdata')
gene_up= mdn[mdn$change == 'UP','ENTREZID']
gene_down=mdn[mdn$change == 'DOWN','ENTREZID']
gene_diff=c(gene_up,gene_down)
gene_all=as.character(mdn[ ,'ENTREZID'] )
## KEGG pathway analysis
### 做KEGG数据集超几何分布检验分析,重点在结果的可视化及生物学意义的理解。
if(F){
### over-representation test
kk.up <- enrichKEGG(gene = gene_up,
organism = 'hsa',
universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
head(kk.up)[,1:6]
dotplot(kk.up );ggsave('kk.up.dotplot.png')
kk.down <- enrichKEGG(gene = gene_down,
organism = 'hsa',
universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
head(kk.down)[,1:6]
dotplot(kk.down );ggsave('kk.down.dotplot.png')
kk.diff <- enrichKEGG(gene = gene_diff,
organism = 'hsa',
pvalueCutoff = 0.05)
head(kk.diff)[,1:6]
dotplot(kk.diff );ggsave('kk.diff.dotplot.png')
kegg_diff_dt <- as.data.frame(kk.diff)
kegg_down_dt <- as.data.frame(kk.down)
kegg_up_dt <- as.data.frame(kk.up)
down_kegg<-kegg_down_dt[kegg_down_dt$pvalue<0.05,];down_kegg$group=-1
up_kegg<-kegg_up_dt[kegg_up_dt$pvalue<0.05,];up_kegg$group=1
source('functions.R')
g_kegg=kegg_plot(up_kegg,down_kegg)
print(g_kegg)
ggsave(g_kegg,filename = 'kegg_up_down.png')
# ### GSEA
# kk_gse <- gseKEGG(geneList = geneList,
# organism = 'hsa',
# nPerm = 1000,
# minGSSize = 120,
# pvalueCutoff = 0.9,
# verbose = FALSE)
# head(kk_gse)[,1:6]
# gseaplot(kk_gse, geneSetID = rownames(kk_gse[1,]))
#
# down_kegg<-kk_gse[kk_gse$pvalue<0.05 & kk_gse$enrichmentScore < 0,];down_kegg$group=-1
# up_kegg<-kk_gse[kk_gse$pvalue<0.05 & kk_gse$enrichmentScore > 0,];up_kegg$group=1
#
# g_kegg=kegg_plot(up_kegg,down_kegg)
# print(g_kegg)
# ggsave(g_kegg,filename = 'kegg_up_down_gsea.png')
#
#
}
### GO database analysis
### 做GO数据集超几何分布检验分析,重点在结果的可视化及生物学意义的理解。
library(org.Hs.eg.db)
{
g_list=list(gene_up=gene_up,
gene_down=gene_down,
gene_diff=gene_diff)
if(T){
go_enrich_results <- lapply( g_list , function(gene) {
lapply( c('BP','MF','CC') , function(ont) {
cat(paste('Now process ',ont ))
ego <- enrichGO(gene = gene,
universe = gene_all,
OrgDb = org.Hs.eg.db,
ont = ont ,
pAdjustMethod = "BH",
pvalueCutoff = 0.99,
qvalueCutoff = 0.99,
readable = TRUE)
print( head(ego) )
return(ego)
})
})
save(go_enrich_results,file = 'go_enrich_results.Rdata')
}
}
class(go_enrich_results)
#下面这部分在后面会一一再结合出图显示
dotplot(go_enrich_results$gene_up[[1]])
dotplot(go_enrich_results$gene_up[[2]])
dotplot(go_enrich_results$gene_up[[3]])
gene_up_CC <- as.data.frame(go_enrich_results$gene_up[[3]])
dotplot(go_enrich_results$gene_down[[1]])
gene_down_MF <- as.data.frame(go_enrich_results$gene_down[[1]])
dotplot(go_enrich_results$gene_down[[2]])
dotplot(go_enrich_results$gene_down[[3]])
gene_down_CC <- as.data.frame(go_enrich_results$gene_down[[3]])
获得的kegg图如下,是将kegg_up和kegg_down在一张图片中展示的,但是此处的红色和蓝色并不是同后面的dotplot图的红色、蓝色的意义一样,后面的红色代表p值小,蓝色代表p值大,而下图中的红色仅仅是代表上面一步在做enrichKEGG后,又对得到的kk.down和kk.up进行了pvalue<0.05的筛选 ,得到的down_kegg和up_kegg合起来作出的图,所以红色代表上调基因富集到哪些通路,蓝色代表下调基因富集到哪些通路。
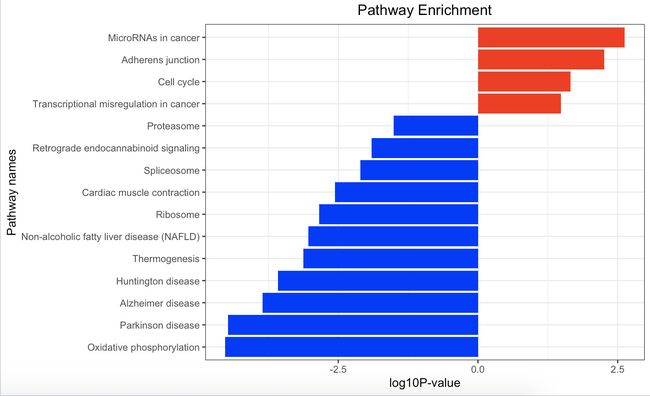
结下来在kegg数据库中提取基因,就是在down_kegg和up_kegg这两个数据框中提取基因。
down_kegg
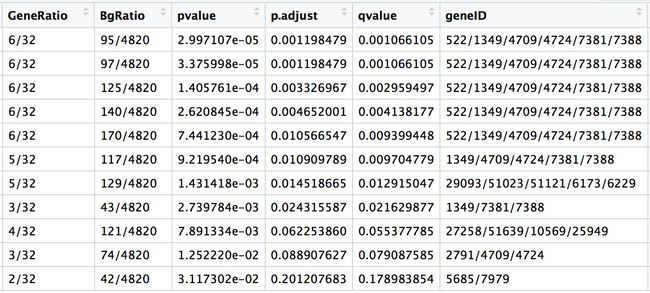
> x <- as.data.frame(down_kegg$geneID)
> apply(x,1,function(x){str_split(x,'/',simplify = T)})
[[1]]
[,1] [,2] [,3] [,4] [,5] [,6]
[1,] "522" "1349" "4709" "4724" "7381" "7388"
[[2]]
[,1] [,2] [,3] [,4] [,5] [,6]
[1,] "522" "1349" "4709" "4724" "7381" "7388"
[[3]]
[,1] [,2] [,3] [,4] [,5] [,6]
[1,] "522" "1349" "4709" "4724" "7381" "7388"
[[4]]
[,1] [,2] [,3] [,4] [,5] [,6]
[1,] "522" "1349" "4709" "4724" "7381" "7388"
[[5]]
[,1] [,2] [,3] [,4] [,5] [,6]
[1,] "522" "1349" "4709" "4724" "7381" "7388"
[[6]]
[,1] [,2] [,3] [,4] [,5]
[1,] "1349" "4709" "4724" "7381" "7388"
[[7]]
[,1] [,2] [,3] [,4] [,5]
[1,] "29093" "51023" "51121" "6173" "6229"
[[8]]
[,1] [,2] [,3]
[1,] "1349" "7381" "7388"
[[9]]
[,1] [,2] [,3] [,4]
[1,] "27258" "51639" "10569" "25949"
[[10]]
[,1] [,2] [,3]
[1,] "2791" "4709" "4724"
[[11]]
[,1] [,2]
[1,] "5685" "7979"
> lapply(x,function(x){str_split(x,'/',simplify = T)})
$`down_kegg$geneID`
[,1] [,2] [,3] [,4] [,5] [,6]
[1,] "522" "1349" "4709" "4724" "7381" "7388"
[2,] "522" "1349" "4709" "4724" "7381" "7388"
[3,] "522" "1349" "4709" "4724" "7381" "7388"
[4,] "522" "1349" "4709" "4724" "7381" "7388"
[5,] "522" "1349" "4709" "4724" "7381" "7388"
[6,] "1349" "4709" "4724" "7381" "7388" ""
[7,] "29093" "51023" "51121" "6173" "6229" ""
[8,] "1349" "7381" "7388" "" "" ""
[9,] "27258" "51639" "10569" "25949" "" ""
[10,] "2791" "4709" "4724" "" "" ""
[11,] "5685" "7979" "" "" "" ""
tmp <- as.data.frame(lapply(x,function(x){str_split(x,'/',simplify = T)})[[1]])
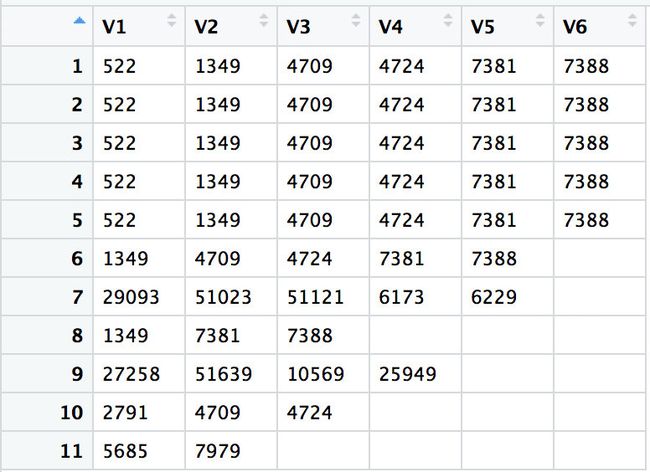
> tmp <- as.data.frame(lapply(x,function(x){str_split(x,'/',simplify = T)})[[1]])
> unique(unlist(apply(tmp,2,unique)))
[1] "522" "1349" "29093" "27258" "2791" "5685" "4709" "51023"
[9] "7381" "51639" "7979" "4724" "51121" "7388" "10569" ""
[17] "6173" "25949" "6229"
> unique(unlist(apply(tmp,2,unique)))[-16]
[1] "522" "1349" "29093" "27258" "2791" "5685" "4709" "51023"
[9] "7381" "51639" "7979" "4724" "51121" "7388" "10569" "6173"
[17] "25949" "6229"
上面得到的是geneID,把这个geneID和mdn的ENTREZID是一个,在这个mdn对应后得到基因名symbol.
tmp1 <- unique(unlist(apply(tmp,2,unique)))[-16]
tmp2 <- mdn[match(tmp1,mdn$ENTREZID),]$symbol
> tmp2
[1] "ATP5PF" "COX7B" "MRPL22" "LSM3" "GNG11" "PSMA4" "NDUFB3"
[8] "MRPS18C" "UQCRB" "SF3B6" "SEM1" "NDUFS4" "RPL26L1" "UQCRH"
[15] "SLU7" "RPL36A" "SYF2" "RPS24"
上面是得到down_kegg的基因
up_kegg

同上面获得down_kegg的步骤差不多,最后得到down_kegg的基因
> tmp <- up_kegg$geneID
> tmp
[1] "994/1786/2033" "2033/5770" "994/2033" "64919/3695"
> str_split(tmp,'/',simplify = T)
[,1] [,2] [,3]
[1,] "994" "1786" "2033"
[2,] "2033" "5770" ""
[3,] "994" "2033" ""
[4,] "64919" "3695" ""
> str_split(tmp,'/')
[[1]]
[1] "994" "1786" "2033"
[[2]]
[1] "2033" "5770"
[[3]]
[1] "994" "2033"
[[4]]
[1] "64919" "3695"
> unlist(str_split(tmp,'/'))
[1] "994" "1786" "2033" "2033" "5770" "994" "2033" "64919" "3695"
> mdn[match(unlist(str_split(tmp,'/')),mdn$ENTREZID),]$symbol
[1] "CDC25B" "DNMT1" "EP300" "EP300" "PTPN1" "CDC25B" "EP300" "BCL11B"
[9] "ITGB7"
> unique(mdn[match(unlist(str_split(tmp,'/')),mdn$ENTREZID),]$symbol)
[1] "CDC25B" "DNMT1" "EP300" "PTPN1" "BCL11B" "ITGB7"
上面是得到up_kegg的基因。
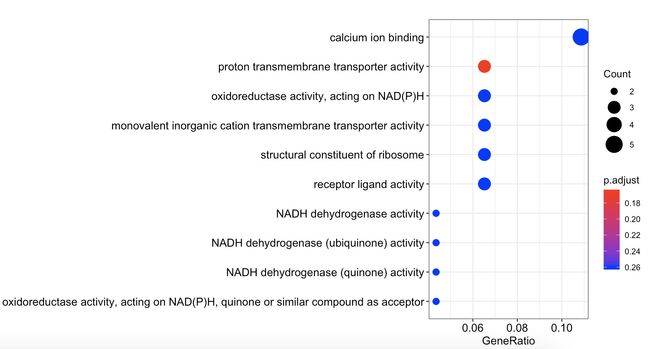
接下来的想法就是gene_up和gene_down在GO数据库中的三种通路中是否富集('BP','MF','CC')分别画气泡图。对于
p.adjust<0.05的某个通路,就新生成个数据框,作为后面提取基因用。
gene_up在GO三种通路中
dotplot(go_enrich_results$gene_up[[1]])
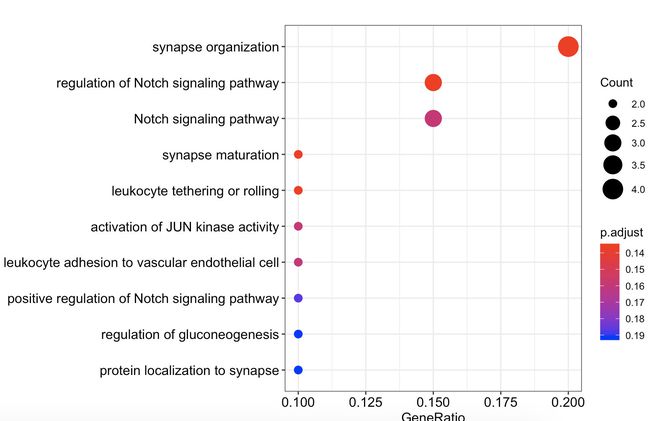
dotplot(go_enrich_results$gene_up[[2]])

dotplot(go_enrich_results$gene_up[[3]])
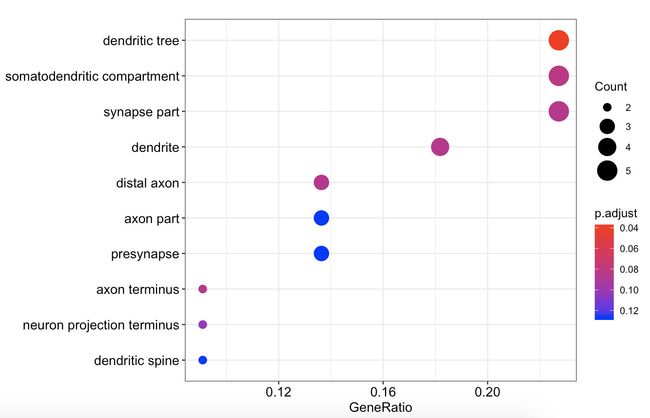
gene_up_CC <- as.data.frame(go_enrich_results$gene_up[[3]])#新生成gene_up_CC数据框
gene_down在GO三种通路中
dotplot(go_enrich_results$gene_down[[1]])
gene_down_MF <- as.data.frame(go_enrich_results$gene_down[[1]])#新生成gene_down_MF数据框
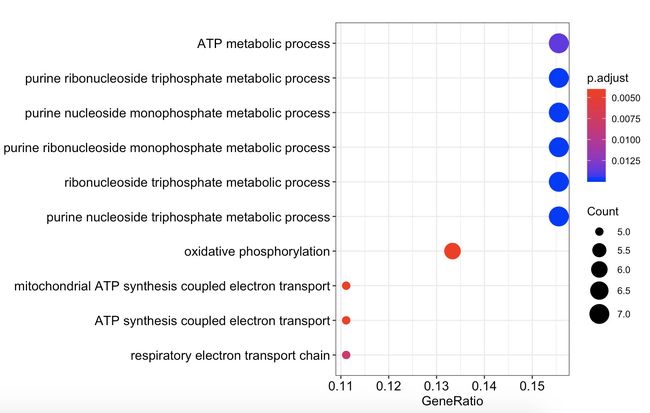
dotplot(go_enrich_results$gene_down[[2]])
dotplot(go_enrich_results$gene_down[[3]])
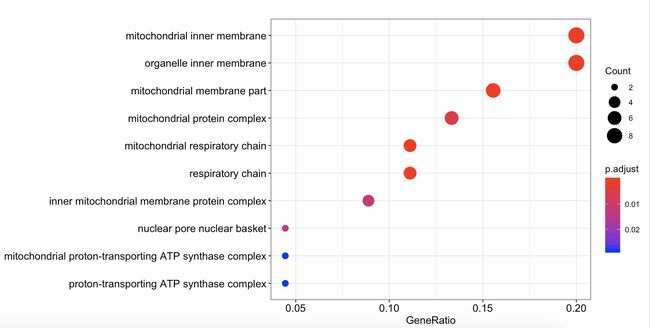
gene_down_CC <- as.data.frame(go_enrich_results$gene_down[[3]])#新生成gene_down_CC数据框
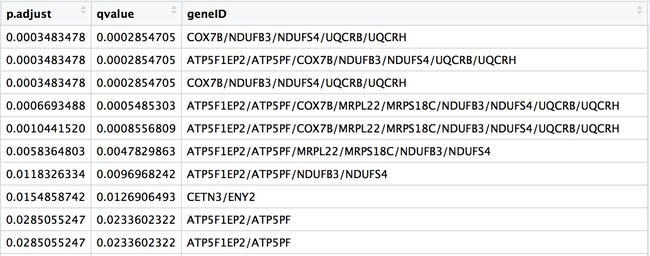
GO通路中富集的基因,以p.adjust<0.5为临界值
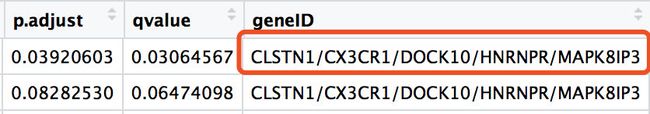
- gene_up_CC的基因获得如下
> gene_up_CC[1,]$geneID
[1] "CLSTN1/CX3CR1/DOCK10/HNRNPR/MAPK8IP3"
> unlist(str_split(gene_up_CC[1,]$geneID,'/'))
[1] "CLSTN1" "CX3CR1" "DOCK10" "HNRNPR" "MAPK8IP3"
- gene_down_MF的基因获得如下
x <- gene_down_MF[gene_down_MF$p.adjust<0.05,]
x1 <- x$geneID
x2 <- str_split(x1,'/',simplify=T)
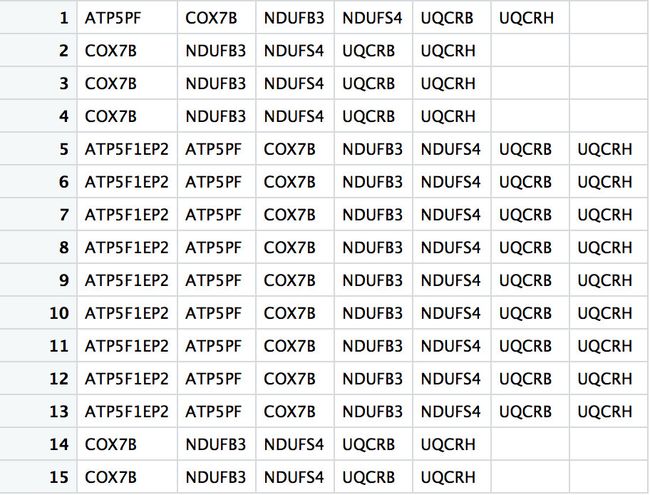
> apply(x2,2,function(x){unique(x)})
[[1]]
[1] "ATP5PF" "COX7B" "ATP5F1EP2"
[[2]]
[1] "COX7B" "NDUFB3" "ATP5PF"
[[3]]
[1] "NDUFB3" "NDUFS4" "COX7B"
[[4]]
[1] "NDUFS4" "UQCRB" "NDUFB3"
[[5]]
[1] "UQCRB" "UQCRH" "NDUFS4"
[[6]]
[1] "UQCRH" "" "UQCRB"
[[7]]
[1] "" "UQCRH"
> unique(lapply(x2,function(x){unique(x)}))
[[1]]
[1] "ATP5PF"
[[2]]
[1] "COX7B"
[[3]]
[1] "ATP5F1EP2"
[[4]]
[1] "NDUFB3"
[[5]]
[1] "NDUFS4"
[[6]]
[1] "UQCRB"
[[7]]
[1] "UQCRH"
[[8]]
[1] ""
> unlist(unique(lapply(x2,function(x){unique(x)})))
[1] "ATP5PF" "COX7B" "ATP5F1EP2" "NDUFB3" "NDUFS4" "UQCRB"
[7] "UQCRH" ""
> unlist(unique(lapply(x2,function(x){unique(x)})))[-8]
[1] "ATP5PF" "COX7B" "ATP5F1EP2" "NDUFB3" "NDUFS4" "UQCRB"
[7] "UQCRH"
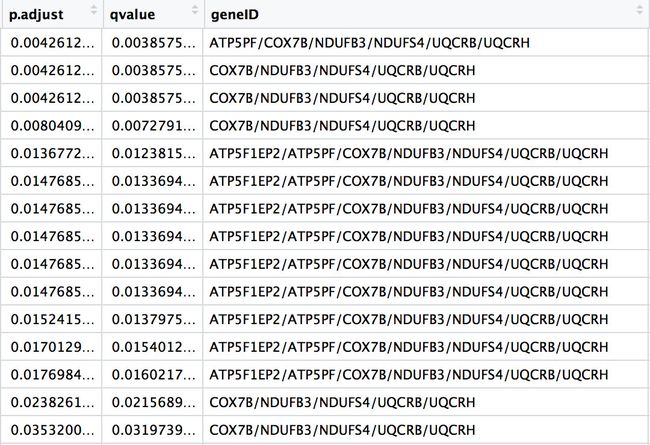
- gene_down_CC的基因获得如下
x <- gene_down_CC[gene_down_CC$p.adjust<0.05,]
x1 <- x$geneID
x2 <- str_split(x1,'/',simplify=T)
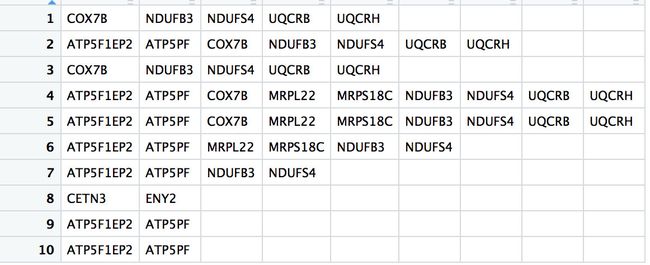
中间过程同上,最后是
> unlist(unique(lapply(x2,function(x){unique(x)})))
[1] "COX7B" "ATP5F1EP2" "CETN3" "NDUFB3" "ATP5PF"
[6] "ENY2" "NDUFS4" "MRPL22" "" "UQCRB"
[11] "MRPS18C" "UQCRH"
> unlist(unique(lapply(x2,function(x){unique(x)})))[-9]
[1] "COX7B" "ATP5F1EP2" "CETN3" "NDUFB3" "ATP5PF"
[6] "ENY2" "NDUFS4" "MRPL22" "UQCRB" "MRPS18C"
[11] "UQCRH"
综上,获得的KEGG和GO的基因如下
KEGG获得的所有富集的基因如下:
[1] "ATP5PF" "COX7B" "MRPL22" "LSM3" "GNG11" "PSMA4" "NDUFB3"
[8] "MRPS18C" "UQCRB" "SF3B6" "SEM1" "NDUFS4" "RPL26L1" "UQCRH"
[15] "SLU7" "RPL36A" "SYF2" "RPS24"
[1] "CDC25B" "DNMT1" "EP300" "PTPN1" "BCL11B" "ITGB7"
GO获得的所有富集的基因如下:
[1] "CLSTN1" "CX3CR1" "DOCK10" "HNRNPR" "MAPK8IP3"
[1] "ATP5PF" "COX7B" "ATP5F1EP2" "NDUFB3" "NDUFS4" "UQCRB"
[7] "UQCRH"
[1] "COX7B" "ATP5F1EP2" "CETN3" "NDUFB3" "ATP5PF"
[6] "ENY2" "NDUFS4" "MRPL22" "UQCRB" "MRPS18C"
[11] "UQCRH"
新建一个.txt文档,长如下图

#读进去R里
a <- read.table('genelist.txt',sep = '\t')
得到如下图

> a1 <-apply(a,1,function(x){gsub('\\[[0-9]*\\]','',x)})
[1] " ATP5PF COX7B MRPL22 LSM3 GNG11 PSMA4 NDUFB3 "
[2] " MRPS18C UQCRB SF3B6 SEM1 NDUFS4 RPL26L1 UQCRH "
[3] " SLU7 RPL36A SYF2 RPS24"
[4] " CDC25B DNMT1 EP300 PTPN1 BCL11B ITGB7 "
[5] " CLSTN1 CX3CR1 DOCK10 HNRNPR MAPK8IP3"
[6] " ATP5PF COX7B ATP5F1EP2 NDUFB3 NDUFS4 UQCRB "
[7] " UQCRH "
[8] " COX7B ATP5F1EP2 CETN3 NDUFB3 ATP5PF "
[9] " ENY2 NDUFS4 MRPL22 UQCRB MRPS18C "
[10] " UQCRH "
a1 <- as.data.frame(a1)

上面得到的图片中的基因可以在继续unique,得到最后的,和原文的比较,目前看好像没有完全一样的,下回分解。