scATAC分析神器ArchR初探-简介(1)
scATAC分析神器ArchR初探-ArchR进行doublet处理(2)
scATAC分析神器ArchR初探-创建ArchRProject(3)
scATAC分析神器ArchR初探-使用ArchR降维(4)
scATAC分析神器ArchR初探--使用ArchR进行聚类(5)
scATAC分析神器ArchR初探-单细胞嵌入(6)
scATAC分析神器ArchR初探-使用ArchR计算基因活性值和标记基因(7)
scATAC分析神器ArchR初探-scRNA-seq确定细胞类型(8)
scATAC分析神器ArchR初探-ArchR中的伪批次重复处理(9)
scATAC分析神器ArchR初探-使用ArchR-peak-calling(10)
scATAC分析神器ArchR初探-使用ArchR识别标记峰(11)
scATAC分析神器ArchR初探-使用ArchR进行主题和功能丰富(12)
scATAC分析神器ArchR初探-利用ArchR丰富ChromVAR偏差(13)
scATAC分析神器ArchR初探-使用ArchR进行足迹(14)
scATAC分析神器ArchR初探-使用ArchR进行整合分析(15)
scATAC分析神器ArchR初探-使用ArchR进行轨迹分析(16)
8-使用scRNA-seq定义簇身份
除了允许通过基因评分分配聚类身份外,ArchR还可以与scRNA-seq整合。这可以帮助进行群集标识分配,因为您可以直接使用在scRNA-seq空间中调用的群集,也可以在整合后使用基因表达测量。整合工作的方式是通过比较scATAC-seq基因得分矩阵和scRNA-seq基因表达矩阵,直接将来自scATAC-seq的细胞与来自scRNA-seq的细胞进行比对。引擎盖下,这种对准是使用执行FindTransferAnchors()从函数Seurat包,可让您跨两个数据集对齐数据。但是,要针对成千上万个单元适当地缩放此过程,ArchR通过将整个单元格划分为较小的单元组并执行单独的对齐方式来提供此过程的并行化。
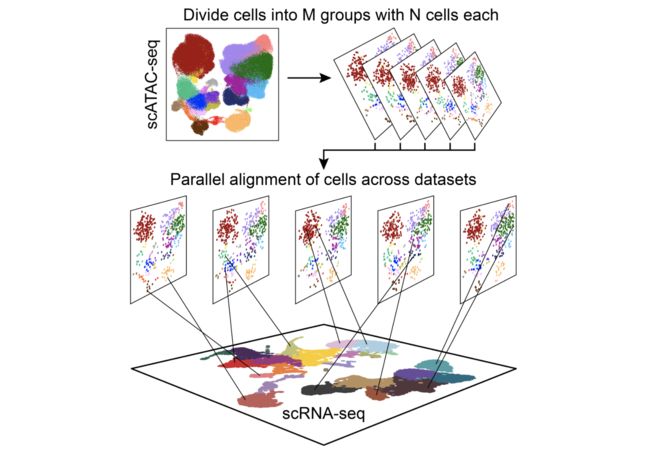
实际上,对于scATAC-seq数据中的每个细胞,此整合过程会在scRNA-seq数据中找到看起来最相似的细胞,并将基因表达数据从该scRNA-seq细胞分配给scATAC-seq细胞。最后,已为scATAC-seq空间中的每个细胞分配了一个基因表达标记,该标记可用于许多下游分析。本章说明了如何使用此信息来分配簇,而后面的章节则说明了如何使用链接的scRNA-seq数据进行更复杂的分析,例如鉴定预测的顺式调控元件。我们相信,随着多组学单细胞谱分析的商业化,这些综合分析将变得越来越重要。
8.1 scATAC-seq细胞与scRNA-seq细胞的跨平台连接
为了将本教程的scATAC-seq数据与匹配的scRNA-seq数据进行整合,我们将使用源自Granja *等人(2019)相同造血细胞类型的scRNA-seq数据。
我们已将此scRNA-seq数据存储为111 MB RangedSummarizedExperiment对象。但是,ArchR还接受未修改的Seurat对象作为集成工作流程的输入。下载并检查此对象后,我们看到它具有基因表达计数矩阵和相关的元数据。
if(!file.exists("scRNA-Hematopoiesis-Granja-2019.rds")){
download.file(
url = "https://jeffgranja.s3.amazonaws.com/ArchR/TestData/scRNA-Hematopoiesis-Granja-2019.rds",
destfile = "scRNA-Hematopoiesis-Granja-2019.rds"
)
}
seRNA <- readRDS("scRNA-Hematopoiesis-Granja-2019.rds")
seRNA
元数据包含一个称为的列BioClassification,其中包含scRNA-seq数据集中每个细胞的细胞类型分类。
colnames(colData(seRNA))
使用table()我们可以看到每个scRNA-seq细胞类型分类中有多少个细胞。
table(colData(seRNA)$BioClassification)
我们可以执行两种类型的集成方法。无限制整合是一种完全不可知的方法,它将获取scATAC-seq实验中的所有细胞,并尝试使其与scRNA-seq实验中的任何细胞对齐。尽管这是可行的初步解决方案,但我们可以通过限制集成过程来提高跨平台对齐的质量。执行受限集成我们使用细胞类型的先验知识来限制比对的搜索空间。例如,如果我们知道scATAC-seq数据中的簇A,B和C对应于3个不同的T细胞簇,并且我们知道scRNA-seq数据中的簇X和Y对应于2个不同的T细胞簇,我们可以告诉ArchR具体尝试将scATAC-seq群集A,B和C中的细胞与scRNA-seq群集X和Y中的细胞进行比对。我们在下面说明了这两种方法,首先对目标的初步群集身份进行无限制的整合,然后然后使用此先验知识执行更精细的约束集成。
8.1.1无约束集成
为了将scATAC-seq与scRNA-seq集成在一起,我们使用了该addGeneIntegrationMatrix()函数。如上所述,此函数通过参数接受一个Seurat对象或一个RangedSummarizedExperiment对象seRNA。我们执行的第一轮集成将是无限制的初步集成,我们不会将其存储在Arrow文件(addToArrow = FALSE)中。我们为积分矩阵提供了一个名称,该名称将ArchRProject通过matrixName参数存储在via中。此功能的其他关键参数提供了将在cellColData其中存储某些信息的列名:nameCell将存储来自匹配的scRNA-seq单元格的单元格ID,nameGroup将存储来自scRNA-seq单元格的组ID,nameScore并将存储跨平台整合分数。
projHeme2 <- addGeneIntegrationMatrix(
ArchRProj = projHeme2,
useMatrix = "GeneScoreMatrix",
matrixName = "GeneIntegrationMatrix",
reducedDims = "IterativeLSI",
seRNA = seRNA,
addToArrow = FALSE,
groupRNA = "BioClassification",
nameCell = "predictedCell_Un",
nameGroup = "predictedGroup_Un",
nameScore = "predictedScore_Un"
)
可以如下所示使用此不受约束的集成,以执行更精细的约束集成。
8.1.2约束集成
现在我们已经有了初步的无约束集成,我们将确定一般的单元格类型以提供一个框架来进一步完善集成结果。
鉴于本教程数据来自造血细胞,我们理想情况下应限制整合以将相似的细胞类型关联在一起。首先,我们将从scRNA-seq数据中找出哪些细胞类型在每个scATAC-seq簇中最丰富。这样做的目的是使用无约束积分来识别scATAC-seq和scRNA-seq数据中与T细胞和NK细胞相对应的细胞,以便我们可以使用此信息进行约束整合。为此,我们将创建一个confusionMatrix,观察一个交叉点,Clusters并predictedGroup_Un包含由scRNA-seq识别的细胞类型。
cM <- as.matrix(confusionMatrix(projHeme2$Clusters, projHeme2$predictedGroup_Un))
preClust <- colnames(cM)[apply(cM, 1 , which.max)]
cbind(preClust, rownames(cM)) #Assignments
上面的列表显示了在12个scATAC-seq簇中的每一个中,哪种scRNA-seq细胞类型最丰富。
首先,让我们从不受约束的整合中使用的scRNA-seq数据查看细胞类型标签:
unique(unique(projHeme2$predictedGroup_Un))
从上面的列表中,我们可以看到scRNA-seq数据中与T细胞和NK细胞相对应的簇是簇19-25。我们将创建这些集群的基于字符串的表示形式,以在下游约束集成中使用。
#From scRNA
cTNK <- paste0(paste0(19:25), collapse="|")
cTNK
然后,我们可以采用所有其他群集,并为所有“非T细胞,非NK细胞”群集(即群集1-18)创建基于字符串的表示形式。
cNonTNK <- paste0(c(paste0("0", 1:9), 10:13, 15:18), collapse="|")
cNonTNK
这些基于字符串的表示形式是模式匹配字符串,我们将使用它们grep来提取与这些scRNA-seq细胞类型相对应的scATAC-seq簇。|字符串中的in作为or语句,因此我们最终在preClust混淆矩阵的列中搜索与模式匹配字符串中提供的scRNA-seq簇编号之一匹配的任何行。
对于T细胞和NK细胞,这标识scATAC-seq簇C7,C8和C9:
#Assign scATAC to these categories
clustTNK <- rownames(cM)[grep(cTNK, preClust)]
clustTNK
对于非T细胞和非NK细胞,这标识了剩余的scATAC-seq簇:
clustNonTNK <- rownames(cM)[grep(cNonTNK, preClust)]
clustNonTNK
然后,我们执行类似的操作以识别对应于这些相同细胞类型的scRNA-seq细胞。首先,我们在scRNA-seq数据中识别T细胞和NK细胞
#RNA get cells in these categories
rnaTNK <- colnames(seRNA)[grep(cTNK, colData(seRNA)$BioClassification)]
head(rnaTNK)
然后,我们在scRNA-seq数据中识别Non-T细胞Non-NK细胞。
rnaNonTNK <- colnames(seRNA)[grep(cNonTNK, colData(seRNA)$BioClassification)]
head(rnaNonTNK)
为了为这种受限的集成做准备,我们创建了一个嵌套列表。这是SimpleList多个SimpleList对象中的一个,我们要约束的每个组一个。在此示例中,我们分为两组:一组称为TNK,标识两个平台上的T细胞和NK细胞,另一组称为,标识两个平台上NonTNK的Non-T细胞和非NK细胞。每个SimpleList对象都有两个单元格ID向量,一个称为ATAC,一个称为RNA,如下所示:
groupList <- SimpleList(
TNK = SimpleList(
ATAC = projHeme2$cellNames[projHeme2$Clusters %in% clustTNK],
RNA = rnaTNK
),
NonTNK = SimpleList(
ATAC = projHeme2$cellNames[projHeme2$Clusters %in% clustNonTNK],
RNA = rnaNonTNK
)
)
We pass this list to the `groupList` parameter of the `addGeneIntegrationMatrix()` function to constrain our integration. Note that, in this case, we are still not adding these results to the Arrow files (`addToArrow = FALSE`). We recommend checking the results of the integration thoroughly against your expectations prior to saving the results in the Arrow files. We illustrate this process in the next section of the book.
#~5 minutes
projHeme2 <- addGeneIntegrationMatrix(
ArchRProj = projHeme2,
useMatrix = "GeneScoreMatrix",
matrixName = "GeneIntegrationMatrix",
reducedDims = "IterativeLSI",
seRNA = seRNA,
addToArrow = FALSE,
groupList = groupList,
groupRNA = "BioClassification",
nameCell = "predictedCell_Co",
nameGroup = "predictedGroup_Co",
nameScore = "predictedScore_Co"
)
8.1.3比较无约束和有约束的积分
为了比较不受约束和受约束的整合,我们将基于通过整合鉴定的scRNA-seq细胞类型对scATAC-seq数据中的细胞进行着色。为此,我们将使用内置的ArchR函数创建一个调色板paletteDiscrete()。
pal <- paletteDiscrete(values = colData(seRNA)$BioClassification)
在ArchR中,调色板本质上是一个命名矢量,其中值是与要与名称关联的颜色相对应的十六进制代码。
pal
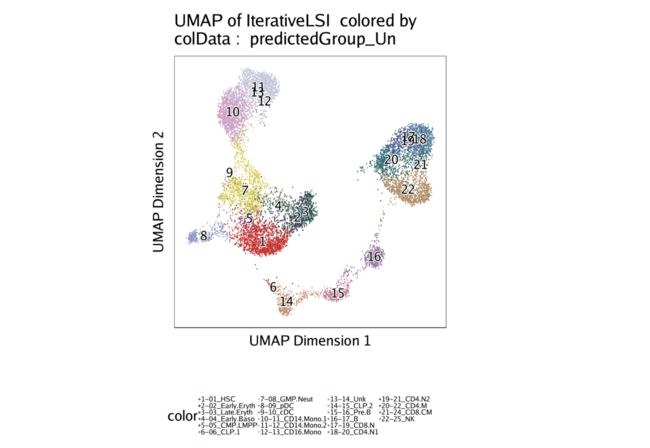
现在,我们可以基于不受约束的整合,通过将scRNA-seq细胞类型覆盖在我们的scATAC-seq数据上来可视化整合。
p1 <- plotEmbedding(
projHeme2,
colorBy = "cellColData",
name = "predictedGroup_Un",
pal = pal
)
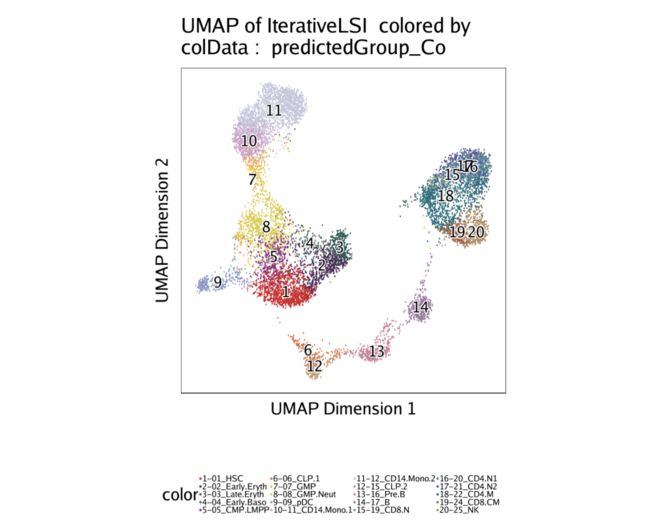
同样,我们可以通过基于约束整合将scRNA-seq细胞类型覆盖在我们的scATAC-seq数据上来可视化整合。
p2 <- plotEmbedding(
projHeme2,
colorBy = "cellColData",
name = "predictedGroup_Co",
pal = pal
)
在此示例中,这些不受约束的集成和受约束的集成之间的差异非常细微,主要是因为目标细胞类型已经非常不同。但是,您应该注意到差异,尤其是在T细胞集群中(集群17-22)。
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(p1,p2, name = "Plot-UMAP-RNA-Integration.pdf", ArchRProj = projHeme2, addDOC = FALSE, width = 5, height = 5)
现在,我们可以projHeme2使用saveArchRProject()函数保存我们的代码。
saveArchRProject(ArchRProj = projHeme2, outputDirectory = "Save-ProjHeme2", load = FALSE)
8.2为每个scATAC-seq单元添加伪scRNA-seq配置文件
现在我们对scATAC-seq和scRNA-seq整合的结果感到满意,我们可以重新运行整合,addToArrow = TRUE将链接的基因表达数据添加到每个Arrow文件中。如前所述,我们通过groupList约束整合和列名nameCell,nameGroup以及nameScore每个元数据列,我们将增加cellColData。在这里,我们创建projHeme3将在本教程中继续进行的操作。
#~5 minutes
projHeme3 <- addGeneIntegrationMatrix(
ArchRProj = projHeme2,
useMatrix = "GeneScoreMatrix",
matrixName = "GeneIntegrationMatrix",
reducedDims = "IterativeLSI",
seRNA = seRNA,
addToArrow = TRUE,
force= TRUE,
groupList = groupList,
groupRNA = "BioClassification",
nameCell = "predictedCell",
nameGroup = "predictedGroup",
nameScore = "predictedScore"
)
现在,当我们使用来检查哪些矩阵可用时getAvailableMatrices(),我们看到GeneIntegrationMatrix已经将其添加到Arrow文件中。
getAvailableMatrices(projHeme3)
有了这个新功能,GeneIntegrationMatrix我们可以将链接的基因表达与通过基因评分获得的推断基因表达进行比较。
首先,确保我们在项目中添加了估算权重:
projHeme3 <- addImputeWeights(projHeme3)
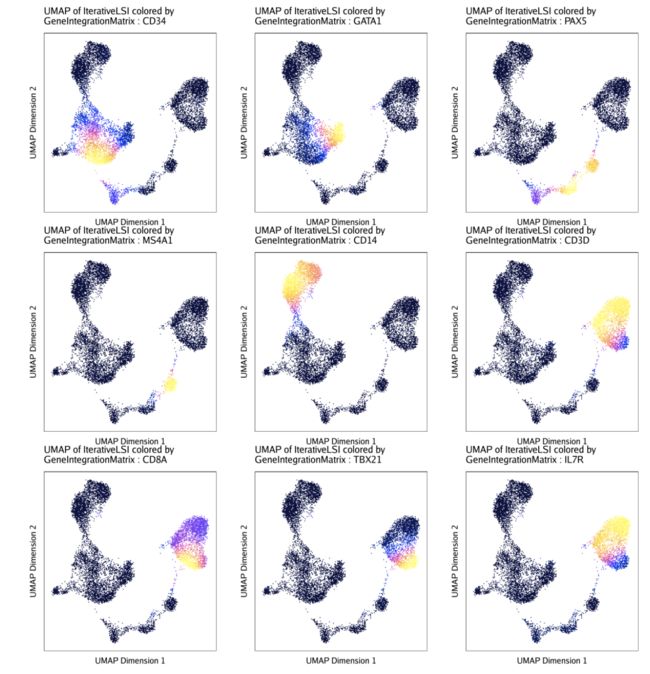
现在,让我们绘制一些UMAP图,上面覆盖我们的基因表达值GeneIntegrationMatrix。
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", #B-Cell Trajectory
"CD14", #Monocytes
"CD3D", "CD8A", "TBX21", "IL7R" #TCells
)
p1 <- plotEmbedding(
ArchRProj = projHeme3,
colorBy = "GeneIntegrationMatrix",
name = markerGenes,
continuousSet = "horizonExtra",
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme3)
)
我们可以制作相同的UMAP图,但将它们与来自我们的基因得分值重叠GeneScoreMatrix
p2 <- plotEmbedding(
ArchRProj = projHeme3,
colorBy = "GeneScoreMatrix",
continuousSet = "horizonExtra",
name = markerGenes,
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme3)
)
我们可以制作相同的UMAP图,但将它们与来自我们的基因得分值重叠GeneScoreMatrix
p2 <- plotEmbedding(
ArchRProj = projHeme3,
colorBy = "GeneScoreMatrix",
continuousSet = "horizonExtra",
name = markerGenes,
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme3)
)
do.call(cowplot::plot_grid, c(list(ncol = 3), p2c))
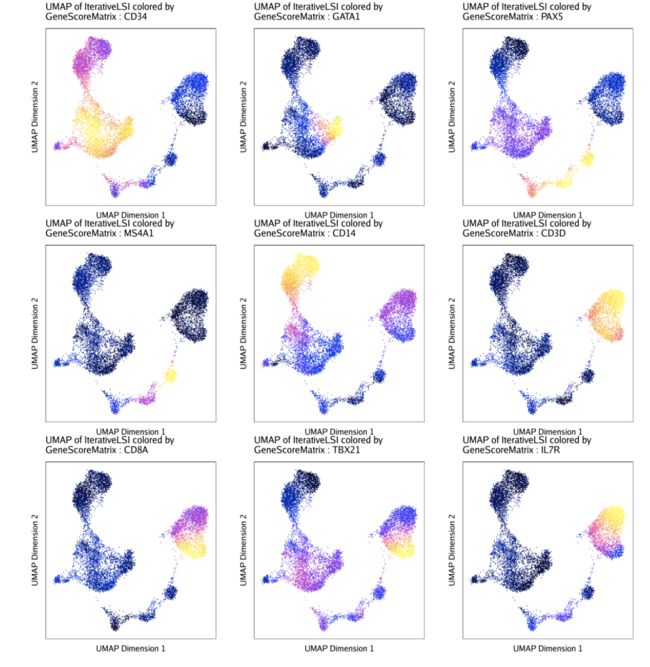
不出所料,这两种推断基因表达的方法的结果相似但不相同。
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(plotList = p1,
name = "Plot-UMAP-Marker-Genes-RNA-W-Imputation.pdf",
ArchRProj = projHeme3,
addDOC = FALSE, width = 5, height = 5)
8.3使用scRNA-seq信息标记scATAC-seq簇
现在,我们对scATAC-seq和scRNA-seq的对齐方式充满信心,我们可以使用scRNA-seq数据中的细胞类型标记scATAC-seq簇。
首先,我们将在我们的scATAC-seq簇和predictedGroup从我们的集成分析中获得的簇之间创建一个混淆矩阵。
cM <- confusionMatrix(projHeme3$Clusters, projHeme3$predictedGroup)
labelOld <- rownames(cM)
labelOld
然后,对于我们的每个scATAC-seq集群,我们确定predictedGroup最能定义该集群的单元类型。
labelNew <- colnames(cM)[apply(cM, 1, which.max)]
labelNew
接下来,我们需要对这些新的群集标签进行重新分类,以创建一个更简单的分类系统。对于每个scRNA-seq簇,我们将其标签重新定义为更易于解释的名称。
remapClust <- c(
"01_HSC" = "Progenitor",
"02_Early.Eryth" = "Erythroid",
"03_Late.Eryth" = "Erythroid",
"04_Early.Baso" = "Basophil",
"05_CMP.LMPP" = "Progenitor",
"06_CLP.1" = "CLP",
"07_GMP" = "GMP",
"08_GMP.Neut" = "GMP",
"09_pDC" = "pDC",
"10_cDC" = "cDC",
"11_CD14.Mono.1" = "Mono",
"12_CD14.Mono.2" = "Mono",
"13_CD16.Mono" = "Mono",
"15_CLP.2" = "CLP",
"16_Pre.B" = "PreB",
"17_B" = "B",
"18_Plasma" = "Plasma",
"19_CD8.N" = "CD8.N",
"20_CD4.N1" = "CD4.N",
"21_CD4.N2" = "CD4.N",
"22_CD4.M" = "CD4.M",
"23_CD8.EM" = "CD8.EM",
"24_CD8.CM" = "CD8.CM",
"25_NK" = "NK"
)
remapClust <- remapClust[names(remapClust) %in% labelNew]
然后,使用该mapLabels()函数,将标签转换为这个新的更简单的系统。
labelNew2 <- mapLabels(labelNew, oldLabels = names(remapClust), newLabels = remapClust)
labelNew2
结合labelsOld和labelsNew2,我们现在可以mapLabels()再次使用该功能在中创建新的群集标签cellColData。
projHeme3$Clusters2 <- mapLabels(projHeme3$Clusters, newLabels = labelNew2, oldLabels = labelOld)
有了这些新标签,我们可以绘制覆盖了新集群标识的UMAP。
p1 <- plotEmbedding(projHeme3, colorBy = "cellColData", name = "Clusters2")
p1
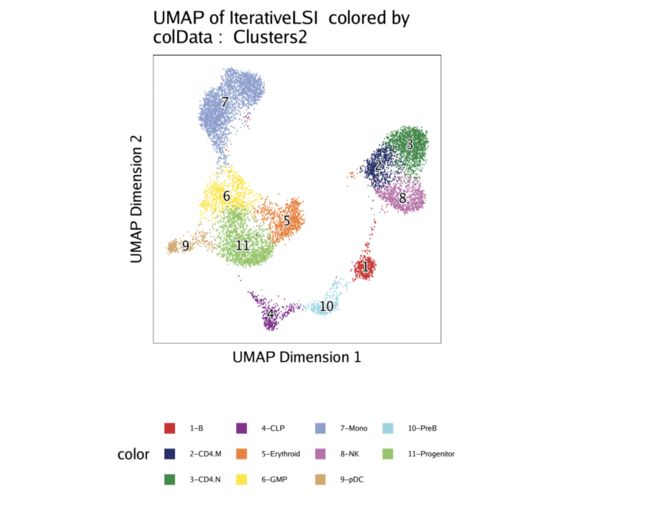
当从已经存在scRNA-seq数据的细胞系统中分析scATAC-seq数据时,此范例非常有用。如前所述,scATAC-seq与scRNA-seq的这种整合还为更复杂的基因调控分析提供了一个漂亮的框架,这将在后面的章节中进行介绍。
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(p1, name = "Plot-UMAP-Remap-Clusters.pdf", ArchRProj = projHeme2, addDOC = FALSE, width = 5, height = 5)
现在,我们可以使用saveArchRProjectArchR 保存原始的projHeme3。
saveArchRProject(ArchRProj = projHeme3, outputDirectory = "Save-ProjHeme3", load = FALSE)
参考材料:
https://www.archrproject.com/