
自然微生物综述(2017 IF:31.851)于2018年5月23日在线发表了Rob Knight亲自撰写(一作兼通讯)的微生物组领域研究方法综述,不仅系统总结了过去,更为未来3-5年内本领域研究方法的选择,提供了清晰的技术路线,让大家走干道,少跳坑,做出更好的研究。值得本领域专业人士细心品读。

Rob Knight (https://knightlab.ucsd.edu/)是谁你还不知道吗?他有多牛呢?仅2017发表高水平文章41篇,截止2018年6月25日,累计发表文章565篇,引用124661次,代表作QIIME引用11215次,h指数高达142 (可怕、恐怖,霍金才62,世界纪录才191)。Rob Knight教授最早在科罗拉多大学任职,目前就职于加州大学圣地亚哥分校微生物组创新中心主任。他是地球微生物组计划(EMP)、美国肠道计划的发起人之一,详见其主页 https://knightlab.ucsd.edu/
复杂的微生物群落形成动态、多变的自然环境,包括哺乳动物肠道、土壤等。DNA测序技术的和数据分析发展极大地推动了微生物组学物种鉴定、假阳性率控制等方面研究。本文作者从实验设计、分子分析技术选择、数据分析方法以及综合多种组学数据集等不同方面,对如何实现最优的微生物组学研究进行探讨。比如对近期快速发展的精确序列变异(exact sequence variants)的方法替代传统基于OTU的聚类分析,将宏基因组学和代谢组学相结合的方法,组成性数据分析等方面进行探讨。值得注意的是,尽管这些方法很新颖,但在研究中还是应当关注实验设计和与研究可重复性相关的经典问题。本综述对这些问题进行了总结,帮助研究者深入了解微生物组数据。
无论是哺乳动物肠道还是深海沉积物,DNA测序技术的快速发展改变了我们对各类复杂生境中微生物群落组成和动态变化的认识。这些技术上的发展推动从临床研究到生物技术等科学领域微生物组研究数量激增。与之而来的是研究人员留下的大量实验数据,并使用一系列令人眼花缭乱的计算工具和方法进行分析。和其他研究一样,在微生物组研究中,扎实的实验是至关重要的,实验方法、环境因素和分析都会影响最终结果。虽然本领域当前研究获得了很多令人信服的成果,但数据收集和分析方法的标准却仍不断变化。
微生物组分析方法和标准在快速发展。特别是过去的两年中,使用精确序列变异来替代OTU分析进行差异丰度检测,以及相关性分析发展迅速。可以预期,在宏基因组分类和功能方面、从多个测序数据中整合数据集、进一步改善机器学习、组成型数据分析以及多种组学分析等其他领域,也有类似的进展。然而,很多与微生物研究相关的基本问题都来自于统计和实验设计问题。因此本领域目前最重要的挑战是,整理微生物组研究独有的新方法,制定可以广泛应用于科学研究的标准。
一篇文章中很难完整涵盖本领域所有内容,本文旨在为微生物组实验设计和分析所得数据提供直接的指导标准,特别关注人类、模式生物以及环境微生物组。
设计可以获得有意义数据的实验是分析的第一步。典型的科学问题,例如病例控制和纵向干预研究等都可以放在微生物组的背景下研究。研究者可以分析在不同群落之间或时间序列下,微生物群落之间结构组成、遗传学或功能的潜在差异。值得注意的是,无论样本来源是什么,微生物组分析的普遍方法(见)都适用,但是,这些分析的特定细节取决于样品来源,例如,从成功的宏基因组测序结果来看,不同样品的16S rRNA基因所需的扩增区域是不一样的。
在评估不同样品时,还需要考虑的重要问题是实验设计和样品收集。对人类微生物组相关研究容易出现的问题进行分析,发现实验设计对研究过程非常重要,通常这些值得注意的问题在动物模型和环境样品中同样适合(见)。
微生物组学分析中,可重复性至关重要。类似的微生物组相关研究常常产生矛盾的结果,如果没有详细的样品采集方法、实验设计、数据处理和分析过程记录就很难检查和解释出现问题的原因。随着本领域新技术的发展,也有必要使用新的工具来重新分析一些早期的实验数据,因为重复性对此类研究非常重要。在收集样品时,采样的详细过程应当完整记录,并且应当考虑到更多的影响因素。另外,实验中要遵循基因组标准联盟提出的标记基因(marker gene)和宏基因组的基因组最小信息标准(minimum information standards, MIxS):MIMARKS和MIMS。这些标准保证各个数据集可以横向比较。在生物信息学处理过程中,研究人员应该跟踪它们运行的所有命令和软件版本,并且将原始数据储存在公共数据库中。我们推荐使用,等工具来实现这个目的,然后将其储存在GitHub等版本控制管理系统中。一些软件包,例如以及等,可以通过整合数据系统自动追踪研究者的这些信息。和是强大的组学分析和数据存档工具,二者结合起来可以使研究者在成千上万的其他样品的大数据背景下分析自己的微生物组数据,同时这些数据也可以被其他研究者再次使用。(这些软件、数据库的简介和链接见文末部分)
尽管,微生物组数据分析方法广泛应用于多种样品类型和环境中,对于不同的样品,实验设计和方法的选择还是需要认真全面的考虑。首先要注意的问题是样品的组成和使用不同方法的可行性。对于被非微生物DNA严重污染的样品,如植物、动物组织(通常宿主DNA占样本的90-99%,想要获得6 Gb微生物数据,理论上需要测序60 - 600 GB原始数据)等如果不排除掉宿主的DNA,鸟枪法宏基因组测序是不太可行的。根据不同的实验问题,如果样品被死亡微生物等DNA遗迹严重污染(如土壤样品),则需要在提取DNA之前使用物理方法来去除遗迹DNA(relic DNA),例如使用单叠氮化丙锭或其他方法。收集的样本量也取决于样本类型,比如生物量较高的粪便样品可能只需要使用拭子、棉棒,而微生物密度较低的样品可能需要较大的体积或浓度才能获取足够的DNA。例如,海洋微生物群落样品通常需要大量的水进行过滤,才能浓缩并获取足够的物质进行DNA提取。尽管这样,在所有情况下,都应当包括合理的控制措施,尤其是需要全面控制取样过程中的污染物,需要维持环境中较低的生物量,例如血液、脊液或者干净的实验室工作环境。实际上,DNA污染物在很多试剂中都能找到,包括拭子、DNA提取试剂盒和PCR试剂。另外,样品的保存方法同样由分析方法和样品类型决定。举个例子,宏转录组需要RNA酶抑制剂,宏代谢组需要保存样品的同时不影响其代谢物的提取和数据收集。
除了考虑样品采集之外,实验设计和原始数据的采集也需要根据样品类型和环境进行仔细调整。例如,动物研究需要评估同笼(co-housing cage)效应,并且应当将实验组分成多个亚类。应当收集新鲜的样品,并且将原始的小鼠情况记录在原始数据中。环境样品则需要收集和环境条件相关的原始数据,如pH、盐度、海拔、取样深度等。收集的方式很大程度上取决于样品类型,在此可能无法对所有的样品进行详细说明。总之,研究中收集、保存和储存的方法应该在所有样品中保持一致,以避免混淆和变异。在室温储藏期间,样品的组分可能会受到某些微生物生长的影响。室温下保存样本方法选择,推荐阅读Microbiome:室温存储样本方法比较。
开展一项可信度高的微生物组研究需要考虑众多因素
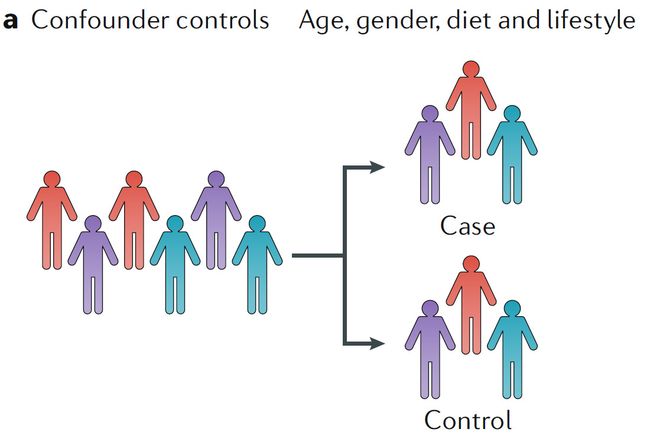
a. 混杂因子对照:年龄、性别、饮食和生活方式
按年龄、性别、饮食和生活方式等潜在的混杂因子分层(分组/分类 stratification)可以部分解决由于混杂效应掩盖组间真实差异的问题
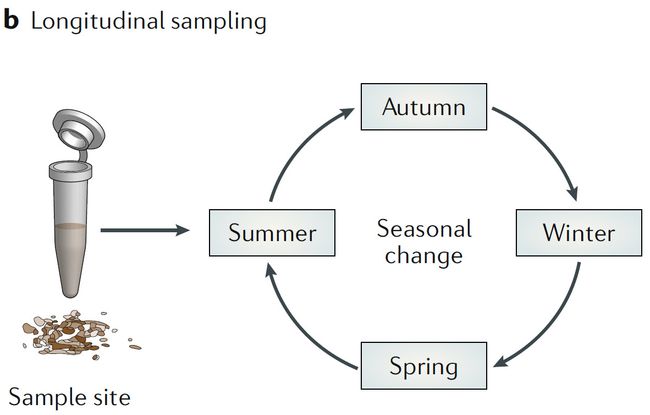
b. 纵向取样
纵向研究是非常有力的手段,即可以控制混杂因子,又可以评估群体的稳定性
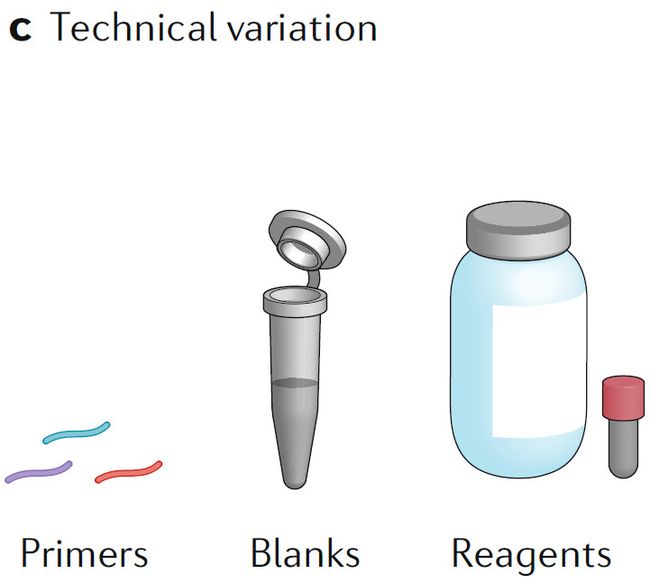
c. 实验技术引入的差异
由于试剂盒、引物、样品储存条件等因素可影响结果,因此实验有标准化的样本处理方法是必须的。需要收集样本处理各阶段的元数据(metadata,相当于样本相关信息),包括临床可变因素、样本处理等,这些对于数据解释非常重要。没有元数据,很难单从测序数据中得出有意义的结论。
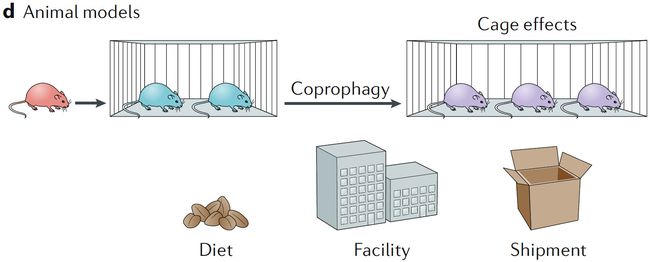
d. 动物模型
动物研究中,食粪性的影响必须在实验设计中注明
对微生物组研究而言,细致的实验设计对获得准确和有意义的结果至关重要。如果不加以控制,很多复杂因素可能会影响和干扰微生物组数据中的一些模式(图1)。认真记录并检查样本信息,合理的对照组(包括提取物、试剂空白对照),周密的实验设计中总体和单个可变因素等都是至关重要的。
首先必须确定实验范围,然后为感兴趣的问题选取适合的实验设计。
例如,横向研究(cross-sectional studies)适用于发现不同人群(如健康个体和疾病患者)或生活在不同区域个体之间的微生物群落差异。然而除了我们所感兴趣的疾病原因之外,个体之间微生物组较大差异的原因也可能是由于饮食、生活习惯以及药物等所致。例如糖尿病患者微生物组变化的研究表明可能与二甲双胍等药物作用相关。而纵向研究(longitudinal studies,),特别是在疾病发作前收集基线样本(baseline samples)可以帮助我们解决这些问题,但此种方法成本较高。为了方便下游统计分析,纵向研究应该仔细规划样品采集的时间安排:对于人类相关研究而言,这可能意味着要为每个被试者在相同的时间点采集样品。有趣的是,与在同一时间点表现出的特定分类群相比,疾病活动的有利预测因子可能更源自于群落的不稳定性。例如和炎症性肠道疾病相比,个体的微生物组群落结构波动比对照组更大。对于包括双盲选随机对照实验在内的介入性(interventional)研究,对于确定治疗过程的微生物组和疾病状态关系中较为有效。
基于分析计划和特定的科学问题来设计实验可以帮助我们确定样本量。(推荐阅读:样品生物学重复数据选择 1必要性 2需要多少重复?)。例如,为了研究新的广谱性抗生素对小鼠肠道菌群的影响,与评估α多样性(定量测定种群内多样性)的变化规律相比,可能需要更多的样本来观察特定类群对抗生药物治疗的影响,因为在不同小鼠间,它们的基础微生物群落组成就是不同的。预期抗生素可以降低所有小鼠的α多样性,但它可能通过不同的方式来影响微生物群落组成。对于任何的实验设计来说,需要采用适当的方法来评估统计能力,辨别技术的可变性以及真实的生物学结果。然而统计能力()和效应量分析()仍然是微生物组研究中的一大挑战。目前用于分析统计能力和效应量分析的方法大多基于相似性分析PERMANOVA(Adonis和ANOSIM方法组间整体差异评估原理)、狄利克雷-多项式分布(Dirichlet Multinomial)或者随机森林分析。随着这些方法的进一步发展,和宏基因组学、宏转录组学、宏蛋白质组学以及代谢组学数据相结合,实验设计和适当样本量的选择也都得到了合理的改进。对于具体的实验设计,建议阅读类似样本类型和预期结果的相关研究。下面我们对微生物组实验设计的一些重要问题进行了扩展。
确定明确的选择和筛选标准,以免混淆新的变量。例如,在个体抗生素治疗后恢复时间的变化表明,在过去六个月内接受抗生素治疗的个体应当排除在微生物组的相关研究之外,类似的,手经过水洗(不用洗手液等清洁剂)后的2个小时后皮肤微生物组才能恢复。
在病例对照实验设计中,必须进行适当的选择和匹配。年龄和性别是最常见的控制标准。但实际上,性别对于大多数人的微生物组而言,在身体各部位的影响较弱,而其他的影响因素,如药物和饮食,相对于其他变量而言往往是更重要的控制因素。这些微生物组变量的相对效应值仍在持续出现中。收集全面的临床数据对于识别无法控制的复杂因素而言至关重要,这个主题的讨论详见15年我发表的综述(McDonald, D., Birmingham, A. & Knight, R. Context and the human microbiome. Microbiome)。对于环境类研究而言,也必须说明类似的混杂因素,在生态学文献中,绘图变化是一个容易混淆的现象,应当使用巢式/嵌套统计检验(nested statistical tests)来解决这个问题。
研究微生物组的主要动物模型是啮齿动物,如小鼠。其他具有不同微生物复杂性的模型,如鱿鱼,昆虫或斑马鱼,通常可用于研究宿主和微生物之间的特定相互作用(例如,微生物群和宿主遗传学如何相互影响)。但是小鼠通常是首选,因为它们具有较好的代表性,并且和人类有较多生理上的相似性。啮齿类微生物组研究需要仔细的实验设计,由于他们具有嗜粪性,因此随着时间的推移,在一个生存空间中的生物学个体和微生物组会变得均匀化,因此实验必须在多个笼子中加以控制,才能防止同笼效应(cage effects)。其他的如母体效应(需要随机化母体效应),避免一只小鼠一只笼子导致的。
即使是基因相同的啮齿动物,由于环境因素(包括饮食,胎次,供应商,运输和设施等)的不同,它们的微生物群体也可能不同。此外,早期微生物组的暴露大大影响已形成的微生物群体,并且有可能影响免疫系统的发育。类似的问题也存在于其他两栖动物模型,如斑马鱼等。
从DNA提取到测序,不同实验方法之间的技术差异很大。在研究中所有样品必须使用相同的试剂盒,并且在纵向研究中,应当收集多个基础样品用来评估时间点的内在变异性。在采样、DNA提取、PCR和测序过程中,设计空白(阴性)对照对于监测污染至关重要。在运输过程中产生的污染的微生物的在分析过程中应当尽量减少,因此样品应当在-80℃保存。对于一些现场研究或其他不能及时冷冻保存的情况,可以使用环境储存方法,例如95%乙醇储存,或商业产品如RNAlater或OMNIgene Gut试剂盒。人工模拟的菌群(Mock communities 具有己知的样品组成成分)可用于标准化分析,即在每次DNA测序过程中包括相同的标准样本。总之,使用不同方法产生的微生物组数据一致依然是一个未能解决的难题。
根据实验的规模(包括整体实验设计,样品类型和来源,测序方法以及下文讨论的其他因素),研究人员可以先获得样本在群落水平上的概述,再进一步从微生物群体组水平对功能变异进行深入的分析的探索。
包括标记基因、宏基因组以及宏转录组测序,不同的微生物组具有不同的方法,从而产生不同的结果。所有广泛应用的方法都具有其不同的优缺点,因此,问题、假设、样品类型和分析目标都应该与所选的方法相关()。在这里,我们对标记基因、宏基因组以及宏转录组的测序成本、合理性、分辨率、以及难度等多方面进行综合比较。概述了二中每个方法的最佳工作流程。如果实验目的是想获到微生物组较高水平、但低分辨率较低的概述,首选标记基因测序。宏基因组测序可以通过分析样品中的总DNA而获得更多的细节,可以在菌株的水平上加以辨别,并提供更多的分子功能信息的基因。对于宏转录组测序,则是更多地用于描述微生物群落中的基因表达。
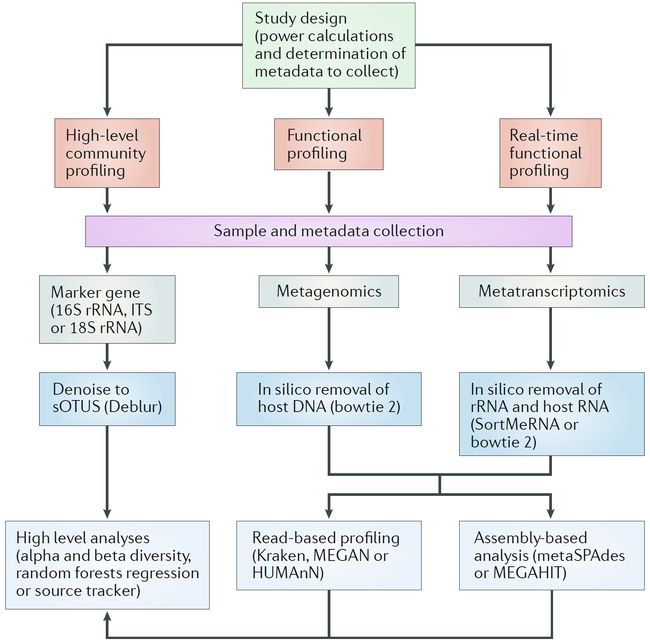
在仔细设计和样本采集后,微生物组数据产生主要包括16S、宏基因组或宏转录组测序。16S测序后,我们推荐使用Deblur获得单碱基变异的参考序列(sOTUs)。尽管DADA2与Deblur结果类似,但Deblur支持并行处理速度更快且更稳定(在不同样品中获得相同sOTUs)。宏基因组和宏转录组首先要去除宿主DNA和RNA,所有rRNA。过滤后的序列可以采用有参方法Kraken、MEGAN或HUMAnN,或De novo组装的方法metaSPAdes和MEGAHIT分析。基于以上三种方法的基本分析,接下来的高级分析,如α, β多样性,物种组成、机器学习等可进一步挖掘微生物组变异的样式。随机森林回归有许多成功的应用,如尸体死亡时间预测,微生物组成熟指数等。来源贝叶斯估计软件SourceTracker可非常有效地估计微生物样本分类在环境中的来源。ITS,转录间隔区。
标记基因测序使用的引物,常常是针对某一感兴趣的特定区域进行设计,从而能够确定样品中微生物的系统发育关系。这个区域通常包含高度可变区,可用于区分研究对象的组成,并且两侧包含可以用作PCR引物结合位点的高度保守区。例如用于细菌和古细菌鉴定的16S rRNA基因和用于真菌鉴定的转录间隔区(ITS)。标记基因的扩增和测序经过了大量的测试,是一种可以高效低成本获得较低分辨率微生物群落结构的方法。这种方法适合于被宿主DNA污染的样品,比如植物或动物组织、以及较低生物量的样品。但是由于这些引物扩增区域的DNA序列不同,可能对DNA序列的亲和力不同产生偏好性,从而影响PCR扩增结果。标记基因测序中的偏好性来源可能是由于不同的可变区选择、扩增子片段大小和PCR循环次数等。引物偏好性对较低生物量的样品影响尤其显著,因为随着PCR次数增多,污染微生物就会被过多的扩增,从而产生较大的影响。优化引物有助于减轻引物偏好,但这需要有关微生物群落组成的一些先验知识,用于评估目标群落中微生物组成分、分类以及覆盖度等。然而,即使经过较好优化的引物也常常受限于种属等分类学水平。标记基因测序通常与基因组背景的相关性较好,所以这也适用于最广泛的样品类型和实验设计。关于扩增子引物选择,可进一步阅读:16S结构 16S单V4区是最佳选择? 引物评估等文章。
宏基因组分析就是对样本内所有微生物基因组进行测序的方法。宏基因组测序与单独的标记基因测序相比,能够获到更加详细的基因组信息以及更高的分类学分辨率,但是在样品制备、测序和分析的成本上更加昂贵。研究者需要得到样品中存在的所有DNA ,包括真核生物DNA以及病毒等。达到足够的测序深度(即每个样品测序读段的数量)、才能够确定物种或者菌株水平的分类学信息、以及尽可能依靠较短的DNA序列来组装成整个微生物基因组。然而,在这种环境下,功能基因的重新注解是不可能实现的。宏基因组测序在基因水平上获得整个群落功能的能力远超标记基因可以分析的范围。但是在文库构建、组装以及参考数据库进行注释等方面,则不如标记基因的方法成熟。随着宏基因组领域的发展,这些注释步骤将得到进一步的验证和改进,关于宏基因组学的全面综述,推荐阅读2017年自然生物技术的综述:宏基因组从取样到分析(Quince, C., Walker, A. W. & Simpson, J. T. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844 (2017).)。
宏转录组分析是通过使用RNA测序来分析微生物组的转录过程,从而提供关于基因表达和微生物组功能活性等信息。之前介绍的标记基因以及宏基因组写个装方法,仅对样品中的DNA序列进行分析,不管其细胞存活情况和活性如何,而宏转录组是以RNA为研究对象。虽然有一些方法从死细胞中消除遗迹DNA,但对微生物RNA进行测序可以更好地了解微生物群落的功能活性,但对于转录活性较高的生物体有一定的偏向。值得注意的是,采用叠氮溴化丙锭(propidium monoazide, PMA)去除遗迹DNA的方法也是获得活性微生物组的可选方法之一。宿主RNA污染,特别是较高丰度的rRNAs,也是另一个重要的考虑因素,应当考虑从样本中去除rRNAs的方法。尽管有些样品类型可能有专门的RNA纯化方案,RNA还是必须小心保存,以免在各种情况下被降解。例如,土壤样品需要去除酶抑制腐殖质。尽管这些技术较为困难,但是转化成宏转录组数据可以为研究者提供新颖独特的见解;转录组的变化幅度要大于宏基因组,宏转录组可以研究微生物群落对异型生物质(药物、杀虫剂、致癌物等)的响应过程。如果你想全面了解宏转录组学分析,请阅读《使用宏转录组进行微生物组研究》的文章(Bashiardes, S., Zilberman- Schapira, G. & Elinav, E. Use of metatranscriptomics in microbiome research. Bioinform. Biol. Insights. 10, 19–25 (2016).)。
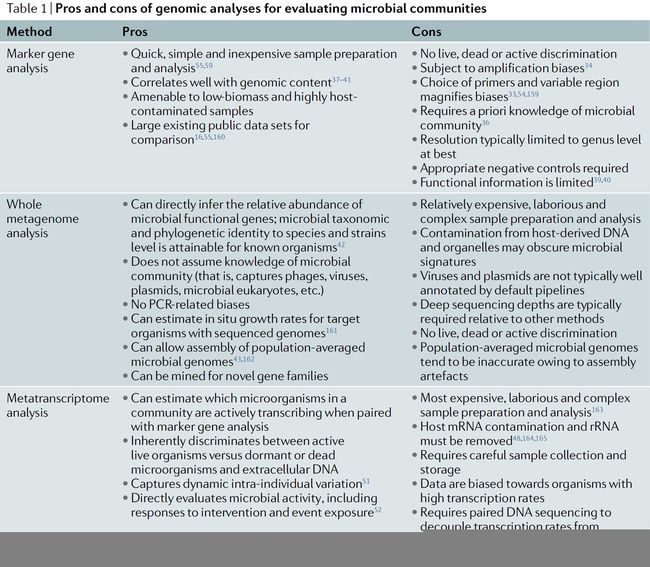
标记基因分析
优点
- 样品制备和分析速度快、简单、成本较低
- 与基因组含量的相关性较高,适合于生物量较低、宿主DNA污染程度较高的样品
- 可用于与现有的大量公共数据集比较
缺点
- 不能区分DNA来源中生物体是否有生命
- 受到扩增偏好性的影响较大
- 引物和可变区的选择对结果影响较大
- 要求对微生物群落有一定的先验知识
- 物种鉴定分辨率通常限于属水平
- 需要适当的阴性对照
- 获得的功能信息有限
全宏基因组测序
优点
- 可以直接获得微生物功能基因的相对丰度;鉴定分辨率可达物种、甚至菌株水平
- 不需要微生物群落相关的先验知识(如捕获噬菌体、病毒、质粒以及微小真核生物等)
- 一般不会产生PCR偏好性
- 可以估算有参考基因组微生物的原位生长速率
- 可组装获得群体平均基因组(甚至可以获得其中一些微生物较完整的基因组)
- 可以挖掘新的基因家族
缺点
- 成本相对较高,样品制备和分析较复杂
- 来自宿主和细胞器的DNA污染可能会掩盖微生物的特征
- 病毒和质粒通常无法自动化注释
- 与其他方法相比,通常需要较高的测序通量(几G - 几百G)
- 不能区分DNA来源于有生命或无生命的生物体
- 由于受组装影响,平均群体微生物基因组往往不准确
全宏转录组测序
优点
- 当与标记基因分析结合使用时,可以估算群落中哪些微生物正在进行积极的转录过程
- 只能鉴定活动生物,排除休眠、死亡微生物及胞外DNA
- 能够捕捉个体内部的动态变化
- 直接评估微生物的活性,包括对干扰或者暴露等情况的响应
缺点
- 费用最高,样品制备和分析过程最复杂
- 必须排除宿主的mRNA、和rRNA污染
- 样品的收集和存储要十分小心
- 数据结果对有高转录率的生物体有偏向性
- 需要与DNA测序结果结合,才能获得细菌丰度变化和转录率
理想情况下,每个微生物组研究将使用以上三种方法来分析样本,然而在大多数情况下,没有足够的样品信息或足够的项目资金来完成全部三种分析,并且在一些情况下,样品可能并不适用于其中的一种测序方法。因此需要研究人员根据科学问题来选择那种方法是最有效的。如果没有预算限制,我们推荐使用宏基因组学测序,不要使用标记基因测序。然而通常情况下通过标记基因测序可低成本快速获得对微生物群落组成的最基本信息。接下来就取决于研究的重点,研究人员可以继续进行宏基因组学和转录组学测序,但是有可能需要二次研究,进行更合理的样品采集和处理。
综上,标记基因的方法对诸如引物选择之类的技术因素较为敏感,因此应当对实验方案进行充分的验证,例如,在地球微生物组项目中,设置多样化样品统一的实验方案是值得学习的。分析标记基因数据的第一步是去除序列错误:尽管序列错误率很低,在Illumina测序中,每个核苷酸的错误率仅为 ~ 0.1%,但是很大部分明显的序列多样性来源于测序错误。直到最近,这个问题得在序列聚类成OTUs中被发现并关注。OTUs聚类,即将相似的序列(通常具有97%相似性阈值)合并归为单个的小分类单元,然后将序列的变体(包括通过序列错误引入的序列变体)合并成可用于随后分析的单个OTU。但是这种方法会在一定程度上,遗漏一些细微但真实的生物序列变异情况,例如存在SNP的序列本该为多个独立OTUs却被的合并成了单个OTU。基于16S rRNA基因测序中位置的特异性信息,来鉴定单碱基变异(SNP)从而加以区分密切相关但也不同的分类群。诸如和等算法,使用测序错误校正的模型来分析测序数据并将其根据精确序列特征(标记基因序列)分为了亚-OTUs(sOTUs)。这些方法得到的结果是一个DNA序列表,是每个样品中的不同序列数,而不是OTU群组。因此我们推荐,当需要与常见的全长数据参考数据库比对的时候,这些方法替代现有基于OTU的方法,除非需要组合使用不同技术(即Illumina测序和454焦磷酸测序)产生的测序数据或者是引物不同。
一个关键的分析步骤是为微生物序列进行物种分类注释。物种分类常用机器学习的方法,如,分类器,它使用的是传统的贝叶斯模型,在属的水平上,对核苷酸的出现频率进行训练,然后在属的水平上进行分配,准确度可达~80%。另外,较为常见的微生物组分析软件流程还有以及,包括物种分类的功能模块。原则上,与三大参考数据库(三个最具特色且经常使用的是,和)精确匹配应当提供更好的分类学依据,或指定特异性,但是鉴于大量未知的分类群,这种方法的敏感性较差。此外,由片段较短的标记基因构建的系统发育树通常结果较差,将标记基因序列插入到基于全长序列的参考序列系统发育树中是一种更好的做法。另外,应当对未分类的微生物进行核糖核酸序列分析是否为细胞器的序列,如叶绿体、线粒体。在很多研究中,这些细胞器序列是应该在分析前过滤去除的(肠道样品研究中,这些序列可以用来鉴定食用的食物种类,不应当完全忽略)。
功能预测分析是一种将标记基因和可用的微生物基因组相联系的技术,用来预测宏基因组,从而推断其生物功能。这种分析通常需要基于参考数据库生成OTU表,然后基于演绎模型(如)为这些基因含量预测提供置信区间,即在距离参考基因组较远的树置信度低,而在许多参考基因组可用的区域则置信度高。因此,影响这些结果准确性的重要因素就是参考基因组的可用性。预测功能分析的另一个限制就是,有些细菌家族的表型和基因型上存在差异,但是它们的16S rRNA可变区非常相似,难以区分。
大多数可应用于微生物组标记基因测序的统计方法,也同样适合于在接下来高级分析中提到的其它组学数据分析。
研究测序样本的完整核酸情况,可以获得微生物群体更大范围的物种组成、功能和进化方面的信息,甚至污染都可以提供重要的发现(如宿主所占比例,潜在的污染源等)。和扩增子分析类似,分析方法的选择需要考虑样本的来源和特定的假设。这里我们将讨论此类分析的最优方法。
将未组装的DNA或mRNA序列与参考数据库比对,可以获得物种和功能基因注释。随着输入数据和数据量前所未有的增长,为提高分类速度,相关方法也在不断优化。许多工具使用k-mers分类DNA片段的物种,如Kraken;或使用Burrows-Wheeler变换算法实现压缩合并数据库相似序列,如Bowtie2和Centrifuge等软件。关于更广泛的工具选择,我们推荐读者阅读17年基因组生物学的相关软件评测文章(McIntyre, A. B. R. et al. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol.)。物种分类标记基因方法采用广泛关注的单拷贝基因,如MetaPhlAn2一条命令获得宏基因组物种组成, TIPP。此外HUMAnN2:人类微生物组统一代谢网络分析2可进一步注释基因和代谢通路。一些工具整理了功能和物种注释,如MEGAN。因为每条测序序列/读段(reads)是独立的,基于有参比对(read-based)方法对于土壤微生物组的大数据集是有效的。值得注意的是,基于序列相似有参比对的物种和功能注释,数据库的选择是至关重要的。为了更好的描述人类肠道环境的特征,高质量(curated是指是由专业人士校正并审核)的基因组数据库如RefSeq,和蛋白家族数据库如Pfam或UniRef,可以增加结果的准确性并减少计算资源的消耗。对于研究较少的环境样本,可以考虑使用NCBI nr/nt和IMG/M的大数据库,虽然会增加计算资源的消耗和降低物种分类的特异性,但数据库更大结果会更全面无偏。专用数据库用于注释特别的物种和功能类别,如专注噬菌体的PHASTER、抗生素抗性基因的Resfams、环境样本的FOAM。此外,许多宏基因组是有参考基因集的,如Tarar的 海洋样本、华大基因BGI的小鼠肠道样本、MetaHit的人类肠道样本。
另一种分析宏基因组和宏转录组的方法是拼接短序列为长序列(contigs也叫叠连群),这些长序列可进一步按相似性进行分类或分箱(bin按序列组分类物种),以获得部分或完整的微生物基因组。此方法不仅可以挖掘数据的物种和功能基因组成,而且可以预测多基因的生物合成通路,甚至可以使用如antiSMASH:微生物次生代谢物基因簇预测的工具来重构代谢产物的基因簇。
然而,使用基于组装的分析方法是条件的(不适合所有项目),如果样本生物多样性高、存在较多相关菌株、以及测序量覆盖度较低等,会导致低丰度物种在下游分析中不准确或丢失。例如,土壤样本因其微生物多样性较高、物种分布不均匀等特点,组装非常困难(一些研究单样本测序量可达300 Gb)。想要避免复杂的宏基因组组装,可选同行发表的己组装好的宏基因组参考数据集,或组装宏转录组,这样可发现高质量数据集中缺少的“微生物暗物质”。组装推荐的工具有metaSPAdes、MEGAHIT和评估quast。对这些工具的讨论,推荐阅读 17年的宏基因组组装软件评估(Vollmers, J., Wiegand, S. & Kaster, A. K. Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective - not only size matters! PLoS ONE)。
想要组装获得部分或完善的单菌基因组,长序列通常采用MaxBin2,或CONCOCT进行分箱(binning,或分类)为假定的单菌基因组,分箱主要原理是基于丰度和核酸组成等信息。分箱的理论可阅读一文读懂宏基因组binning;实战可阅读分箱宏基因组binning, MaxBin, MetaBin, VizBin。评估分箱基因组的质量,CheckM使用单拷贝基因来估算基因组的完整性和污染率。VizBin可以在不基于参考序列条件下,可视化宏基因组序列组装结果,使用户可以方便查看相关物种的序列分类簇,输助评估分箱的质量。
由于宏基因组组装的复杂性,我们推荐使用在这方面整合好的工作流程,可以自动化进行数据分析,如组装assembly和分箱bin结果可视化分析平台—Anvi’o,ATLAS,或MetAMOS。
为了比较不同测序量的样品,可通过许多标准化方法解决这一问题。常用的标准化方法有RPM (reads per million,每百万的序列数,即百万比,类似于百分比),TPKM (transcripts per kilobase million,每百万单位kb长度转录本数量,对数据量和基因长度同时标准化,使不同基因间相对丰度可比),或相对丰度(relative abundance,如百分比,或总体为1的小数)。此外,有许多工具可以进行更为复杂的标准化方法,如edgeR和DESeq2(采用基于负二项分析的标准化方法,在测序数据领域应用极广泛,edgeR使用实战详见3热图:差异菌、OTU及功能)。
新工具在有参(reads-based)和无参/组装(aseembly-based)方法均快速发展。软件方法的选择、优缺点评估应该基于背景研究清楚的数据集,或人工合成的数据集(Nat. Methods: 宏基因组软件评估—人工重组宏基因组基准数据集),这样才能根据自己的项目特点,选择合适的方法,有利于微生物群体研究获得更合理的结果。
微生物组数据经过处理,可以获得特征(features,如物种不同分类级或基因)与样本的丰度矩阵。但这一结果是存在迷惑性(deceptively)的,因为微生物组数据通常是高维数据,包括几千个不同物种,矩阵数据(表格)稀疏存在许多零值;因此需要注意的统计处理方法,以挖掘有意义的结果。
和多样性常用于评估微生物组的整体变异。Alpha多样性可以量化样品内的特征多样性,也可以进行样品组间比较。例如,我们一个疾病个体与健康对照 ,研究者可比较组间Alpha多样性的物种均值。Alpha多样性物种测量的方法有三类:丰富度(richness)的测量常用观测的物种数(Observed OTU / Richness)和Chao1丰度估计(估计真实物种多样性),进化距离测量采用信任系统发育多样性(Faith’s phylogenetic diversity),这两类方法受样本测序深度影响很大;此外还有一类即考虑丰富度,又考虑均匀度的Shannon指数,对测序量不敏感。详见箱线图:Alpha多样性解读。请注意,这些方法仅限用于16S数据,应用于其它微生物组数据类型可能并不合适。
Beta多样性比较每对样品间特异的差异,产生所有成对样品间的距离矩阵。度量标准的选择对结果影响较大,需牢记我们在解析生物学数据。Bray-Curtis, Canberra, 有权重的UniFrac等定量度量标准采用特征的丰度信息进行计算,binary-Jaccard,无权重的UniFrac定性方法仅考虑特征的有无。进化方法的Unifrac分析需要进化树文件,提供更生物学的解析,但缺少树文件时无法使用。
可提供alpha, beta多样性分析的软件有QIIME、Mothur和R语言vegan包。无参数的置换(permutation)检验方法PERMANOVA、ANOSIM用于估计的不同组间beta多样性的显著性,其中PERMANOVA应用于组间变异较大的数据集更好用。计算Alpha和beta多样性,需要研究者掌握抽样技术(即每个样本中抽取相同数量的序列),不同的抽样数量级可影响结果。目前计算Unifrac最好的方式是稀疏/稀释(rarefracation),但一些特殊情况下的成对差异丰度比较需要完整的样本数据集。
Beta多样性数据可视化采用排序的技术,常用如主坐标轴分析(PCoA)或主成分分析(PCA)。点我读懂PCA和PCoA。这类方法将复杂的距离矩阵,转换为可观察的2或3维空间,代表样品间距离。样品可以按分组信息着色,方便观察组间差异,属于无监督的方法。EMPeror框架提供可交互式的显示PCoA图。
另一种常用分析方法是比较感兴趣组间(处理、对照)微生物或功能(基因、通路)的差异。微生物组数据具有高维、松散组成等特征,鉴定解析微生物群落差异的分类群是极有挑战性的。组成是问题的关键;当一种微生物增长,因为比例总和为1,其它必然会降低。例如,己知某个病人的药物只影响一个微生物属,对其它菌无任何影响。尽量其它微生物不受药物影响,但它们相对丰度减少,是由单个微生物属过度生长引起的。这种情况影响许多经典方法的结果,如参数统计检验(如student’ t-test和ANOVA)、计算相关性(如Spearman排序相关系数)通常导致完全不可接受的超90%假阳性率。最近,组成意识(compositionally aware)方法提到了组成和相对丰度方法的问题。一种方法是在统计检验上强制进行强生物假设:如Lovell’s比例度量方法仅检测正相关。其它一些工具为微生物组数据专门做了优化,假定小部分物种是相关的,大多数的相关系数为0,如SparCC和SPEIC-EASI。BAnOCC是另一个提出组成问题的工作,它对数据无任何假设。我们推荐使用另一种方法,等距对数比例转换(isometric log ratio transform , ilr),用于检测微生物群体间差异。ilr方法控制假阳性率,采用检测微生物丰度对数变化检验,通常认为平衡。平衡构建基于先验知识,如进化历史或微生物对环境因子pH响应的生态位分化。ilr应用后,标准统计工具(如多元响应、线性回归和分类)可更有效的检测平衡或对数比例的微生物组数据差异。最近也有绝对定量的方法,包括测序和细胞计数。
机器学习是在微生物组领域非常有效的方法,可基于当前状态区分样品(分类,由己知的分类与结果学习,预测末知分类,如健康和疾病),或预测将来某一状态。例如,可根据口腔菌群预测牙龈炎的易感性和严重程度。随机森森回归有许多应用,如预测尸体死亡时间、确定儿童菌群成熟度。SourceTracker可以估计末知群体微生物来源和组成,最有用的是可根据环境样品来分类微生物的来源。注意,机器学习需要足够的样本量,用于交叉验证,一定是独立的实验或数据集来确定模型的可靠性。
了解微生物群落的组成并不是研究的终点,我们更想知道群体的功能。多组学数据整合,扩增子测序,宏基因组,宏转录组,宏蛋白组,宏代谢组和其它技术都可用于特定微生物群体功能和组成的深入理解研究。例如,改变的代谢组成反应生物合成的活性,mRNA和蛋白表达,以及蛋白活性。多组学分析将化学和生物学知识结合,提供研究对象更完整的生物学系统的新方法,是一个活跃的研究领域(图3)。

分子生物学的中心法则
以细菌细胞为例:从DNA —— RNA —— 蛋白 —— 代谢物的过程的概述,正好对应多组学研究的6个层面。
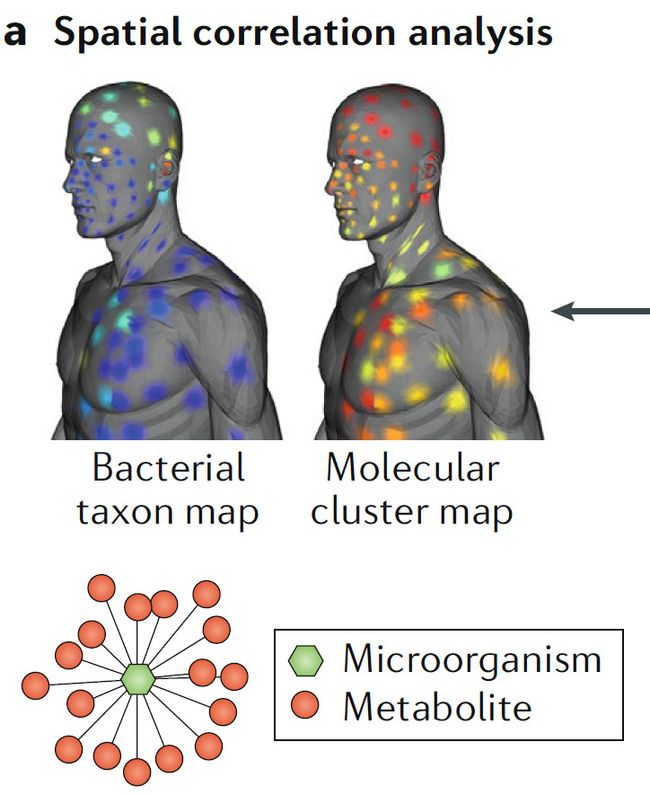
a. 空间相关性分析
采用三维可视化分子和微生物特征地图,帮助我们理解空间相关性
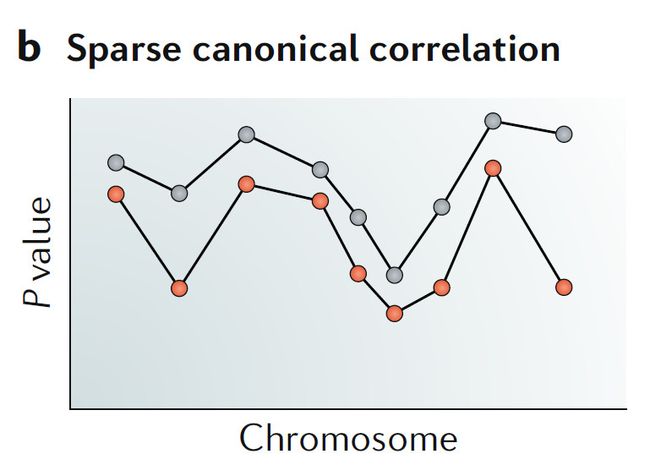
b. 稀疏典型相关分析
鉴定线性的两个子集存在高度相关
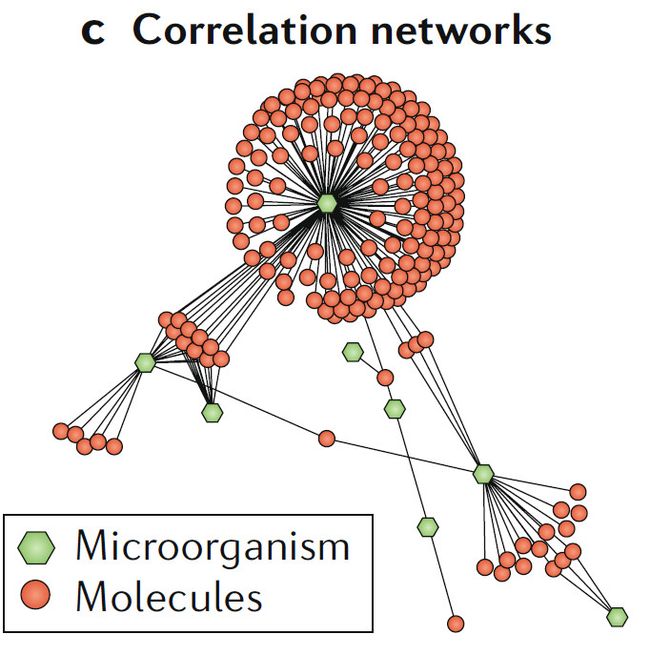
c. 相关网络
相关网络分析展示成簇的微生物与代谢物,这些代谢物可能是相关微生物的产物,方便确定合成源头
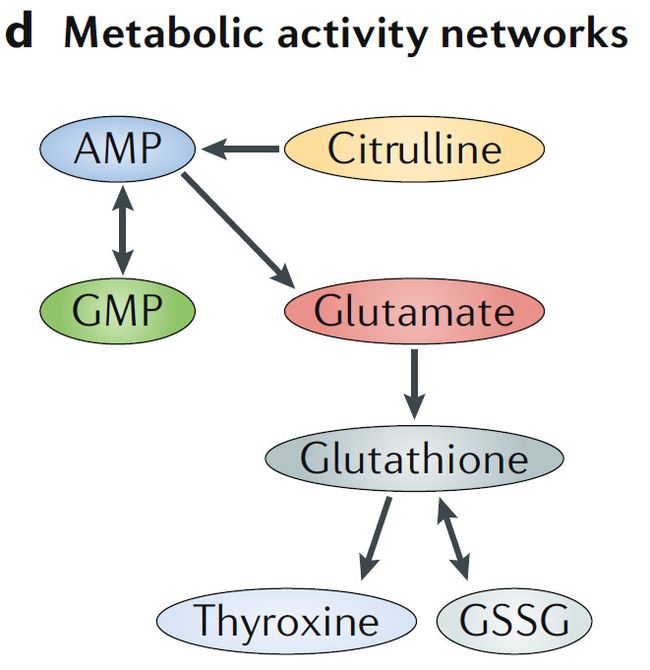
d. 代谢活性网络
依赖特定物种分子机制的数学模型,代谢活性网络帮助预测微生物群体结构和功能
GSSG,氧化型谷胱甘肽
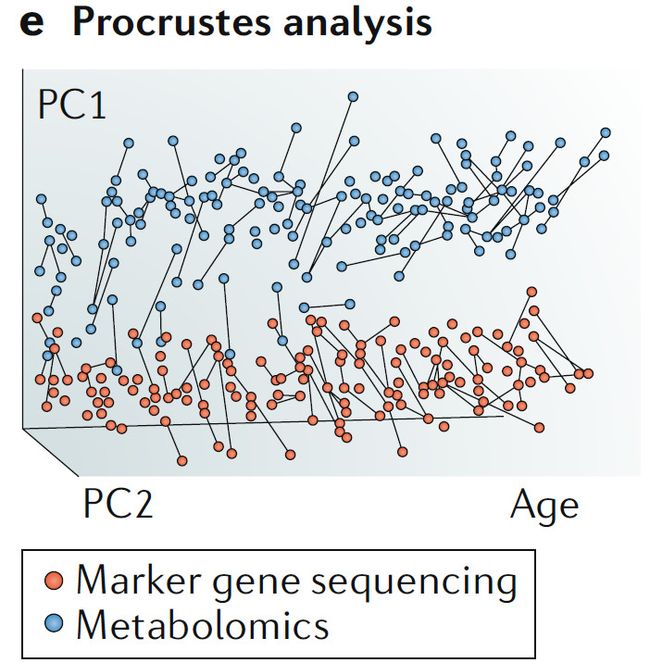
e. 普氏分析法
普氏分析法可以在同一主坐标轴内可视化数据的趋势,直接比较具有相同内部结构的不同组学数据,
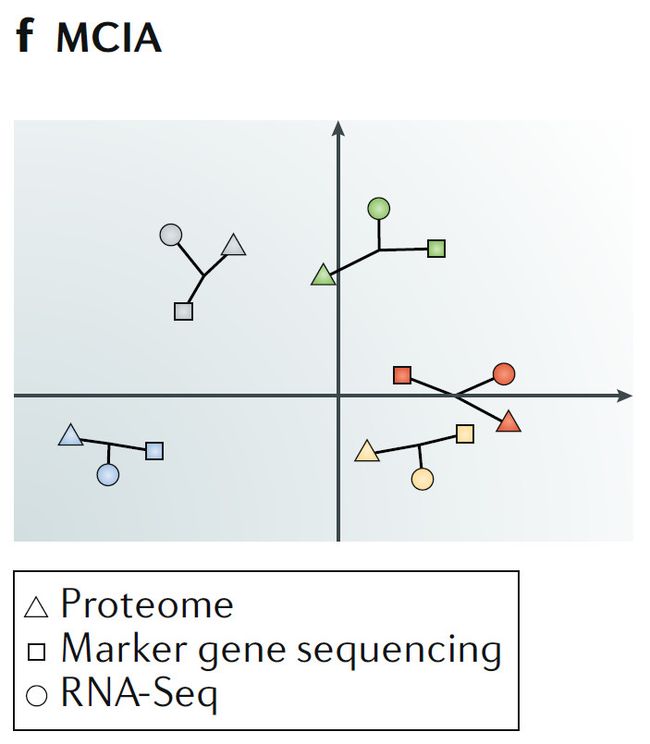
f. 多重共惯性分析
MCIA可以通过图形代表不同类型,多维比较不同组学数据,相似的组学数据可以更容易理解。
RNA-Seq,转录组测序或RNA测序
微生物产生代谢物可影响宿主,微生物群体动态变化并与宿主疾病和健康有关。bread的音标代谢物有益处(如短链脂肪酸)和坏处(基因毒率大肠杆菌素)影响宿主。然而,鉴定微生物组中代谢物来源是非常有挑战的。更有挑战的是鉴定代谢物来源的微生物,收集微生物产物,修饰特定代谢物。下面简单总结解决这些困难的策略:
- 比较自然样品与微生物组培养菌(分离的微生物)代谢物。一种有效的方法是比对临床或环境样品串联质谱和分离培养菌的数据,发现特异的代谢物标志可被认为来源于某个可培养微生物。
- 在微生物基因组和宏基因组中定位代谢物基因。一些代谢物只存在于特定的微生物分类中。检测自然样本的代谢物,可以确定挖掘基因组数据的可能来源。例如,2,3-丁二酮是一种特异的发酵产物,由链球菌代谢产物。检测临床样品,结合生物合成基因可加速定位生物途径至来源物种的基因组。
- 构建微生物与代谢物的共现网络。共现网络或相关方法把微生物与代谢建立联系。这是一个热门研究领域,可用的算法对检测松散的微生物数据进行了优化,如SparCC、CCLasso和其它等。需要注意,此方法在多元数据集中假阳性率过高。
- 无菌与特异无病原小鼠模型。这样比较鉴定微生物组代谢物来定殖小鼠,和末定殖的小鼠。无菌小鼠(包括单菌定殖,定殖群体)有助于鉴定特定的微生物和关注的代谢产物。
整合多组学数据存在本质的困难。例如,基因表达与代谢物来自不同的时间尺度, 微生物产生许多种代谢物,通常仅是响应其它物种的信号。宏基因组和宏代谢组的数据集(数据矩阵中大多数为0)比宏蛋白组的数据更松散,这对很多分析方法无法处理。尽管多组学整合是正在发展中的领域,相关可用工具也逐渐增加。例如XCMS可在线整合代谢物数据和代谢通路,也可整合蛋白组和转录组。传统的成对相关分析方法Spearman和Pearson,也可以进行多组学分析。然而,高维度、高稀疏度的微生物组数据、代谢组数据存在较高假阳性率。普氏分析采用降维数据样本数据间样式或距离,必须的是相关排序空间而不是个体的特征(使用Mantel或PROcrustes随机检验)。其它方法整合组学数据集时,不仅考虑样本间关系,而且关联样本与特定元数据中关注的分类信息(如检查健康、疾病组,或对照与处理组)。此类方法如多重共惯性分析,在两个不同数据集中对样本相关多维数据进行降维,还有相关元数据(relevant
metadata)、偏最小二乘(partial least-squares)、典型相关分析(canonical correlation analysis)、稳健稀疏(robust sparse)典型相关分析(是一种处理稀疏组学数据的方法)。
优秀的综合分析工具有全球自然产物协会(GNPS)分子网络(鉴定代谢物与注释通路)、普适的系统生物学工具如XCMS在线。多组学研究空间样式研究己久,目前正在增加时间序列上的研究。空间地图可以使用工具展示,使研究多维多组学数据更方便挖掘和解析。
综合分析多组学数据需要多种统计方法。但这些方法在微生物组数据中不是最优的。简单发现组学数据内部的相关是第一步,建立因果联系是下一阶段的挑战。介绍了代谢组学和微生物组数据集联合分析方法,使研究从相关向因果推进。在多组学分析中,多重比较校正问题是关键;数据集可能包括几千种不同的微生物和代谢物,所以会有很多偶然的显著相关。校正显著性检验的方法有假阳性率(如Benjamini–Hochberg校正),更保守的总体错误率(family-wise error)校正(如Bonferroni校正)。使用这些方法校正,对降低多组学分析中假阳性率非常有帮助。
尽管仍存众多挑战,但多组学数据联合分析是非常有前景的。也有一些宏基因组、宏转录组和代谢组成功整合的例子,阐明微生物组中基因调控、微生物与代谢物共现相关。这类研究发现远超单组学研究,如研究肠道细菌代谢异生质,和抗生素诱导的微生物组减少产生艰难梭菌适宜的代谢组环境。相对的,宏蛋白组和微生物组数据是一个新研究领域,成功的案例有鉴定Crohn疾病的生物标记、研究永久冻土层中的微生物蛋白产物。此外,宏蛋白组注释和分析的工具正在开发中。综上所述,综合多组学数据可以更全面的理解微生物组的DNA鉴定、蛋白和代谢物功能,使用研究结果可有指导意义。
本综述讨论了微生物研究全阶段工作的指导,从实验设计、收集储存样品、从测序数据的图形结果中挖掘结果,均对结果与生物学解释产生影响。由于许多实验步骤对生物学结果有影响,因此建立标准化的实验步骤是必须的,这样才可能跨实验联合分析。第一步努力是提出推荐使用最优方法,如国际人类微生物标准、微生物质量控制(MBQC)计划(DNA提取也能发Nature?,NB:实验vs分析,谁对结果影响大)。生物信息分析流程和对照也正向标准化而努力,如使用云平台实现可重计算、公开原始数据和分析源代码实现可重复研究,这些方面的快速发展为微生物组领域结果的一致和可比较成为可能。一个最重要的是引入内参标准的标准化(在生物芯片分析领域中已经非常普遍),使微生物组分析中生物学真实样本可以在系统水平量化。
本文主要关注了群体水平DNA层面的分析,转录组和单细胞测序先进技术也涉及并有一些应用。同时提到要避免在昂贵分析中经常出现的错误,如不合理的样本量和验证,使用最优方法作为标准,样本处理,组成型数据分析,和其它常见的陷阱。使用MBQC和环境微生物组(EMP)中标准化、样式清楚的样品收集新方法,可极大缩短探索新方法意义的时间。
本领域的趋势是向前所末有大数据集、理解流行病学家长期熟知的混杂因子、更重视纵向研究设计等将成为重点。尤其是人、动物模型、体外实验在系统层面和大尺度基础上,从观察研究向干预研究是值得考虑的。标准化方法应用的增长,可以降低噪音和偏好,对微生物领域研究从实验室范围向临床、田间和自然环境的深入提供广泛前景。
- :准确序列变异。在扩增子(标记基因)测序数据分析中,使用测序读短的原始序列代替之前聚类生成的OTUs。此方法的出现是受近几年测序错误纠正算法提高才得以实现,代表方法有Delbur, dada2和unoise3,较OTUs仅有属水平的精度相对,此方法最高可达株水平的单碱基精度,推荐阅读 扩增子分析还聚OTU就真OUT了、
主流非聚类方法dada2,deblur和unoise3介绍与比较 - :可操作分类单元(OTUs),经过比对,通常将一组相似性大于97%的序列定义为一个微生物种群(群体)。推荐阅读:16S测序,你必须认识OTU
- :机器学习,使用算法来学习和预测数据。常见的两种应用是分类(如 Nature:肠道菌群高盐与高血压关系)和回归( 如作者早期Sciences发表的16S+功能预测尸体死亡时间
)。 - :元数据,即样品的信息。在很多研究中通常以表格(矩阵)的形式出现,其中样品名称为行,元数据的各种不同分类、属性为列,如年龄、性别、经纬度、平均月降水量、季节、疾病状态等等。
- :样品组内多样性的描述指数。
- :效应量化分析。指量化分析元数据集中的一些类别(如性别、处理组、测序板等)对菌群的影响程度。
- :标记基因。通常指的是如16S rRNA、18S rRNA以及转录间隔区(ITS)等保守基因,它们具有典型特征包括:可以用来鉴定物种分类单元的高可变区,同时其两端是高保守区域可作为PCR引物的结合位点。
- :统计检验中涉及到的和主效应有关的变量。例如,土壤地块就是测试肥料对土壤微生物群的影响的嵌套因子。
- :食粪性,涉及到粪便的消耗。一些动物物种通过食用粪便,对食物中的植物组织进行二次分解消化。人山人海英语这将导致同笼中的动物肠道菌肠较相似。
- :测序读取的单个DNA片段,可翻译为读段,大家在平时交流更喜欢直接叫reads
- :宏转录组测序一个生物群落中基因转录物的总和。
- :腐殖质,通过有机质的生物降解而产生的。腐殖质是腐殖土壤的主要成分。
- :宏基因组,生物群落中遗传物质的总和,例如,人类肠道样品中的所有微生物的所有遗传物质。
- :在机器学习中使用的简单概率分类器,是基于贝叶斯定理的一个应用,推测两类样品间的独立性。
- :通过DNA测序获得的序列中所有可能的长度为k的序列。
- :样品组间多样性的描述指数。
- :基于系统发育进化树来计算样品多样性的一种α多样性方法。
- :描述群落多样性的一个常见的指标,是一种综合指数,它即包括丰富度(richness),又考虑均匀度(evenness)。
- :假阳性率,进行多重比较时,揭示无效假设检验中I型错误率的方法。
- :等距对数比例转换。使用树作为参考,将比例向量转换为对数比例向量。 计算的对数比率由树内相邻分支之间物种比例的平均对数的差异组成。
- :随机森林回归,是一种使用决策树执行分类的机器学习技术,可以用于学习后预测某事发生时间,如生长阶段,死亡时间等。
- :总体错误率,在执行多个假设检验时,发生一个或多个I型错误的概率。
- EBI (http://www.ebi.ac.uk/)
世界三大生物数据库之一,我们常用它存储和分享宏基因组领域产生的海量数据,实现数据共享、保障结果可重复,以及数据的再利用。有很多特色分析工具,尤其在宏基因组领域的分析平台很有名 https://www.ebi.ac.uk/metagenomics/ - Galaxy (https://usegalaxy.org/)
一种生物信息分析平台的框架,把传统的代码分析包装为网页中图形和菜单,可以更方便无程序语言基础的生物学家使用,但与终端下代码交互相比会损失灵活性。如很多软件为方便大家使用,都布置在galaxy平台上,如KO通路预测PICRUSt、 差异分析LEfSe等。 - GitHub (https://github.com)
世界上最大的代码备份和共享平台,近期刚被微软65亿美无收购。现在文章中分析所占的比重非常大,几十到上百项分析,可涉及成千上万行的代码,如不分享原始代码,文章中的结果仅凭方法部分的描述几乎是无法重复的。GitHub为代码、及中间文件的分享提供了目前最方便的平台。很多顶级文章都分享全部分析代码于此,如 德国Paul组的Nature: 培养组学—高通量细菌分离培养鉴定
、美国Dangl组的Nature:根系菌群参与磷胁迫和免疫的平衡等文章中都提供了Github地址,即可以重复大牛的研究,更是非常好的学习材料。 - Jupyter Notebooks (http://jupyter.org)
一种交互式代码编辑器,可以实现代码、注释、结果和格式混排,方便代码运行和结果展示,是很用Python用户的最爱。 - QIIME 2 (https://qiime2.org)
引用过万次的QIIME软件的最新版本,于2018年正式发布,提供了标准化的格式,可实现更好的标准化分析和可重复计算,快速了解可阅读本平台早期翻译的 QIIME2中文教程-把握分析趋势 。需要注意的是,此软件每月都有较大更新,如下定决心使用此流程,请务必阅读官方最新版本英文教程。 - Qiita (http://qiita.microbio.me)
开源的微生物研究管理平台,可支持多组学、多研究的管理和分析,支持第三方的分析流程。 - R Markdown (https://rmarkdown.rstudio.com/)
Markdown是一种轻量标准语言,可以用纯文本快速实现网页效果(公众号每天的推文大部分用Markdown书写)。其中R markdown版本可将R语言统计绘图过程、结果混排为网页,方便共享分析过程,实现可重复计算,在科学计算领域有很广泛的应用,如 斯坦福大学统计系教授带你玩转微生物组分析其中一篇PNAS就提供了整篇文章所有图表分析的代码、小学英语单词大全讲解和结果混排的R markdown文档,方便同行阅读学习。
一文读懂:Rob Knight手把手指导菌群研究(必读综述)
花开 06-18 热心肠日报
原标题:菌群分析的规范
① 菌群研究和分析方法正高速发展,研究方法标准化、数据共享平台的推广为联合独立项目、完善已有成果提供可能;
② 实验设计需合理设置空白和对照组,并考虑实验动物的习性;
③ 可参考对已知菌群的分析效果,决定采用标志基因组、宏基因组还是宏转录组研究手段和分析方法;
④ 基于序列实际差异的菌群分析方法应逐步代替OTU分析;
⑤ 基于菌群相对丰度的相关性分析容易出现假阳性,需要优化分析方法;
⑥ 多组学数据联合有助于进行全面的、机制性的菌群研究。
主编评语
菌群研究和分析方法日新月异,本文系统性地介绍了菌群研究的实验设计、方法选择和数据分析方式,在列举和比较大量研究方法的同时,指出了目前OTU分析、菌群丰度分析和相关性分析的缺陷,强调数据共享、方法标准化的重要性。文中提及大量最新研究、分析方法和平台,指导作用强,值得专业人士参考。
- 文章主页 https://www.nature.com/articles/s41579-018-0029-9
- PDF下载链接 https://sci-hub.tw/10.1038/s41579-018-0029-9
- 何茂章本文阅读笔记 https://note.youdao.com/share/mobile.html?id=1070e1e78a8e1d7d8b7049f160bbd523&type=note&from=timeline
- 一文读懂:Rob Knight手把手指导菌群研究 https://www.mr-gut.cn/papers/read/1052334830
- 相关技术文档链接来自 宏基因组公众号,ID: meta-genome

秦媛,博士在读。2014年毕业于河北农业大学植物保护专业,2017年于中国林业科学研究院获森林保护硕士学位,现就读于中科院遗传发育所。现己发表论文6篇,专利1项;其中第一作者3篇发表于Biotechnology Advances、Frontiers in Microbiology、Fungal Ecology,累计影响因子17.9。主要研究方向包括根际微生物组结构与功能、宏基因组学分析方法和科研插图绘制。

刘永鑫,博士。2008年毕业于东北农大微生物学专业。2014年中科院遗传发育所获生物信息学博士学位,2016年博士后出站留所工作,任宏基因组学实验室工程师,目前主要研究方向为宏基因组学、数据分析与可重复计算和植物微生物组。发于论文10篇,SCI收录7篇。2017年7月创办“宏基因组”公众号,不到一年关注人数超2万,累计阅读超200万。
宏基因组/微生物组是当今世界科研最热门的研究领域之一,为加强本领域的技术交流与传播,推动中国微生物组计划发展,中科院青年科研人员创立“宏基因组”公众号,目标为打造本领域纯干货技术及思想交流平台。公众号每日推送,内容涉及科研思路、实验和分析技术、文献解读、重要成果报导等。目前经过近一年发展,分享近200篇原创文章,已有21000+小伙伴在这里一起学习了,感兴趣的赶快关注吧。
