移步github
测序原理
共有的特点:
优点:无需前期扩增,不引入偏向性;读长长
缺点:错误率高;
10X Genomics:Illumina二代测序的升级版
10X Genomics,是常规Illumina二代测序的升级版,由于开发出了一套巧妙的Barcoding建库方案,使得Illumina这种短读长二代测序能够得到跨度在30-100Kb的linked reads信息,与二代测序数据相结合,在Scaffold的组装上能够得到媲美三代测序的组装结果

首先将每一条长片段的DNA分配至不同的油滴微粒中,通过专利的GEM建库技术,长片段DNA被切碎成适合测序的大小,并且来源于相同油滴(同一条长片段DNA)的DNA片段,会带上相同的一段DNA序列标记(Barcode),之后在Illumina系统上测序完成后,可以理论上再将来源相同的DNA序列独立拼接,得到原先的长片段DNA序列。
其GC偏好性如何?

10X Genomics技术相对于Illumina来说,有改进,但依旧是个拱形,而PacBio则是无偏倚的均一分布。10X的技术,其Coverage一样是受GC含量影响较大的,那么如果真要应用10X技术,那么必须注意目标DNA的GC含量分布最好能控制在30~70%。
Helicos:tSMS
真正的单分子测序(Helicos True Single Molecule Sequencing)
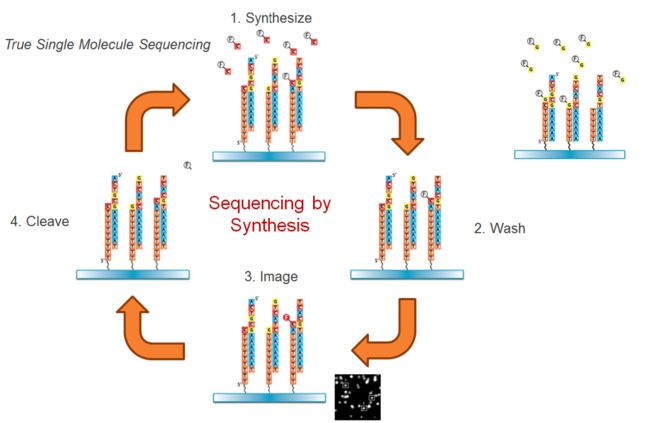
待测DNA 被随机打断成小片段,在每个小片段( 200bp)的末端加上poly-dA,并于玻璃芯片上随机固定多个 poly-dT 引物,其末端皆带有荧光标记,以利于精确定位。
首先,将小片段 DNA 模板与检测芯片上的poly-dT 引物进行杂交并精确定位,然后逐一加入荧光标记的末端终止子。这个终止子与 Illumina 的终止子可不一样,不是四色的,是单色的,也就是说所有终止子都标有同一种染料。
在掺入了单个荧光标记的核苷酸后,洗涤,单色成像,之后切开荧光染料和抑制基团,洗涤,加帽,允许下一个核苷酸的掺入。通过掺入、检测和切除的反复循环,即可实时读取大量序列。最后以软件系统辅助,可分析出完整的核酸序列。
缺点:Heliscope 在面对同聚物时也会遇到一些困难,但可以通过二次测序提高准确度;由于在合成中可能掺有未标记的碱基,因此其最主要的错误来源是缺失。
PacBio:SMRT
PacBio SMRT(single molecule real time sequencing)技术也应用了边合成边测序的思想,并以SMRT 芯片为测序载体。
基本原理是:DNA 聚合酶和模板结合,4 色荧光标记4 种碱基(即是dNTP),在碱基配对阶段,不同碱基的加入,会发出不同光,根据光的波长与峰值可判断进入的碱基类型。
DNA 聚合酶是实现超长读长的关键之一,读长主要跟酶的活性保持有关,它主要受激光对其造成的损伤所影响。
PacBio SMRT 技术的一个关键是怎样将反应信号与周围游离碱基的强大荧光背景区别出来:
它们利用的是ZMW(Zero Mode Waveguide,零模波导孔)原理,如同微波炉壁上可看到的很多密集小孔。小孔直径有考究,如果直径大于微波波长,能量就会在衍射效应的作用下穿透面板而泄露出来,从而与周围小孔相互干扰。如果孔径小于波长,能量不会辐射到周围,而是保持直线状态(光衍射的原理),从而可起保护作用。同理,在一个反应管(SMRT Cell,单分子实时反应孔)中有许多这样的圆形纳米小孔,即ZMW(零模波导孔),外径100 多纳米,比检测激光波长小(数百纳米),激光从底部打上去后不能穿透小孔进入上方溶液区,能量被限制在一个小范围(体积20x10-21L )里,正好足够覆盖需要检测的部分,使得信号仅来自这个小反应区域,孔外过多游离核苷酸单体依然留在黑暗中,从而实现将背景降到最低。

优缺点:
优点:
可以通过检测相邻两个碱基之间的测序时间,来检测一些碱基修饰情况,即如果碱基存在修饰,则通过聚合酶时的速度会减慢,相邻两峰之间的距离增大,可以通过这个来直接检测甲基化等信息
 SMRT can detect base modification
SMRT can detect base modification测序速度很快,每秒约10 个dNTP
读长长
无需PCR扩增,也避免了由此带来的bias
 SMRT features
SMRT features
- 需要的样品量很少,样品制备时间花费少
缺点:
测序错误率比较高(这几乎是目前单分子测序技术的通病),达到15%
 SMRT reads error rate
SMRT reads error rate好在它的出错是随机的,并不会像第二代测序技术那样存在测序错误的偏向,因而可以通过多次测序来进行有效的纠错
Nanopore sequencing
该技术的关键之一是,它们设计了一种特殊的纳米孔,孔内共价结合有分子接头。当DNA 碱基通过纳米孔时,它们使电荷发生变化,从而短暂地影响流过纳米孔的电流强度(每种碱基所影响的电流变化幅度是不同的),灵敏的电子设备检测到这些变化从而鉴定所通过的碱基。
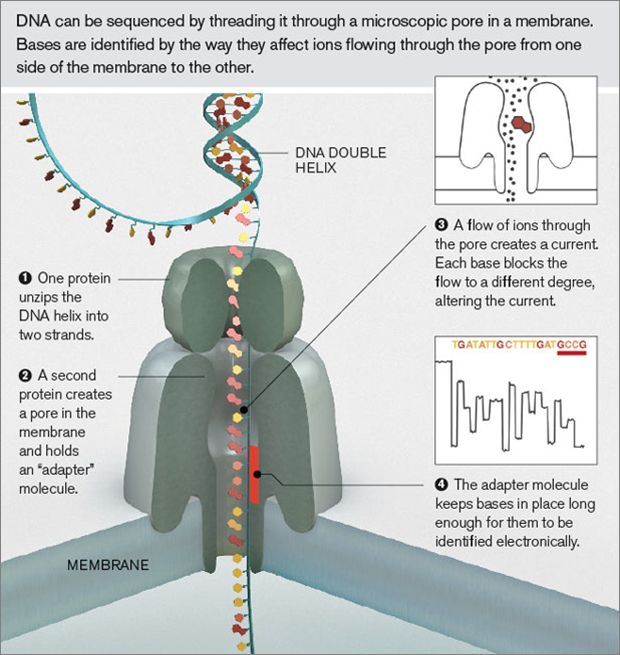
测序原理:
解螺旋,将双链DNA解开成单链。
DNA单链分子通过一个孔道蛋白,孔道中有个充当转换器的蛋白分子。
DNA单分子停留在孔道中,有一些离子通过带来电流变化,而不同的碱基带来的电流变化是不同的。
转化器蛋白分子感受5个碱基的电流变化。
根据电流变化的频谱,应用模式识别算法得到碱基序列。
特点:
- 测序读长
因为测序原理无需要DNA聚合酶的链式反应,所以不存在DNA聚合酶的失活问题,理论上只要DNA分子不断开,就一直可以通过纳米孔,目前在对于人和大肠杆菌的测序种观测到的read是1Mb。
要问测多长,请问您提取的DNA是否够长?
三种不同建库方法Nanopore测序情况
DNA建库方法 序列数 平均读长 Read N50 Ligation Library 451,020 8,012 13,920 Rapid Kit Library 315,684 13,796 30,397 Ultralong reads Protocol 694,659 24,179 99,790 数据说话,Ligation建库方法测序读长的read N50达到14k左右,超长建库方法read N50达到 100k。
- 测序准确率
Nanopore测序准确率和Pacbio持平,为86%左右。而且起始位置正确率偏低,在大约100nt位置达到稳定,且错误为随机测序错误。
 Nanopore position specific error rate
Nanopore position specific error rate如果选择 1D2测序方式,即对于DNA的正负链都进行测序,可以达到96%的准确率
 1D2 reads can imporve correction rate
1D2 reads can imporve correction rate
Nanopore 测序仪 MinION 的一些特征:
1、早期使用基因工程改造过的a-hemolysin蛋白,称为作为biosensor,最新的nanopore使用CsgG 蛋白,它允许ssDNA通过
2、MinION的flow cells中有512 channels,每个channel含有4个pores和sensors,每个channel作为一个独立的测序单元,对一条DNA分子进行测序,DNA分子从四个纳米孔中的一个穿过,产生电流信号。因此一个flow cell可以同时对512条DNA进行测序
3、为了进行dsDNA的测序,需要在dsDNA的两端加上两个接头:leader-adapter 和 hairpin-adapters,且都被预先固定在马达蛋白 (motor proteins) 上
 Nanopore sequencing process
Nanopore sequencing processleader-adapter带着dsDNA到邻近的纳米孔,然后原先固定在leader-adapter的马达蛋白开始将dsDNA打开,使得第一条链,即模板链(template),能够穿过纳米孔,测序过程随即开始
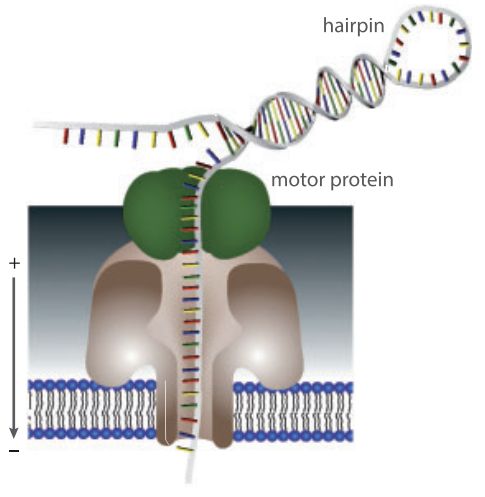 Nanopore sequencing process
Nanopore sequencing process3、MinION的flow cell有多个升级版本(R6.0, R7.0, R7.3, R9 and R9.4),在通量,读长和准确率方面都有很大的提高
 Improvement of Nanopore sequencing quanlity
Improvement of Nanopore sequencing quanlity4、纳米孔中的电流传感器的采样频率为5000 Hz,测序速度为250 bases/s(早期为75 bases/s)
5、目前唯一的便携式DNA和RNA测序仪,注意这里有两个概念,一是便携式,MinION只有100g重,相当于1个大一点的U盘或者小一点的移动电源;二是DNA和RNA测序,和所有NGS测序仪、甚至三代Pacbio不同的是,MinION和其他的ONT仪器们,可以直接对RNA进行测序,无需预先转化为cDNA。此外,一旦启动测序,实时的数据会不断产生,而不用像传统的NGS测序中一个run结束后才能收获数据,一旦数据量足够可随时终止测序进程,简直不要太爽!
ONT公司目前推出的几款测序仪:
- MinION —— flow cell最新版本是R9,内含2048 wells。48h即可产出10~20 Gb数据

- GridION X5 —— 一款桌面式测序仪,通量介于大家熟悉的MinIon和高通量的PromethIon之间。GridIon X5系统一次最多可运行五个MinIon flow cells,可以根据实际数据量的需求一次运行1~5个flow cells。目前的最大通量是,每运行48小时可产出高达100 GB的测序数据。

- PromethION —— 一个具有模块化设计的更大的台式测序仪,其在全功率时的运行能力约为MinION的300倍,通量在Tb级。包括48个flow cells,这些flow cells可以单独运行,也可以一起运行

PacBio-SMRT数据分析
QC
- 下机数据

在analysis文件夹中,下机的数据被分割为三个文件进行存储
- 以bax.h5为后缀的是原始二进制文件;
- 以subreads.fasta / subreads.fastq为后缀的是经一级处理得到的标准格式的碱基文件;
- 以sts.csv / sts.xml为后缀的是记录测序过程中每个ZMW度量指标的统计文件
数据的命名:
m 140415_143853_42175_c100635972550000001823121909121417_s1_p0
└1┘└─────2─────┘ └──3──┘└───────────────4───────────────┘└5┘└6┘
1. m是movie的缩写;
2. 测序时间,格式为yymmdd_hhmmss;
3. 仪器编号;
4. SMRT Cell Barcode;
5和6无实际意义,一般是固定的
- 数据结构
Pacbio 数据的文库模型是两端加接头的哑铃型结构,测序时会环绕着文库进行持续的进行,由此得到的测序片段称为 polymerase reads,即一条含接头的测序序列,其长度由反应酶的活性和上机时间决定。目前,采用最新的 P6-C4 酶,最长的读长可达到 60kb 以上。

polymerase reads 是需要进行一定的处理才能获得用于后续分析的。这个过程首先是去除低质量序列和接头序列:
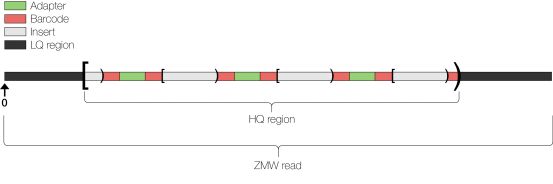
处理后得到的序列称为 subreads,根据不同文库的插入片段长度,subreads 的类型也有所不同。
对长插入片段文库的测序基本是少于2 passes的(pass即环绕测序的次数),得到的reads也称为Continuous Long Reads (CLR),这样的reads测序错误率等同于原始的测序错误率。
而对于全长转录组或全长16s测序,构建的文库插入片段较短,测序会产生多个passes,这时会对多个reads进行一致性校正,得到一个唯一的read,也称为Circular Consensus Sequencing(CCS)Reads,这样的reads测序准确率会有显著的提升。
polymerase reads 与 subreads 是相对应的两个概念
Continuous Long Reads (CLR) 与 Circular Consensus Sequencing(CCS)Reads 是是相对应的两个概念
- 数据质量
不同于二代测序的碱基质量标准Q20/Q30,三代测序由于其随机分布的碱基错误率,其单碱基的准确性不能直接用于衡量数据质量。那么,怎么判断三代测序的数据好不好呢?
-
长度
长度短的测序数据不一定差(与文库大小有关),但差的数据长度一定短。在上游实验环节,最关键的影响因素是文库的构建。高质量的文库产出的数据长度长,质量好;而低质量的文库产出的数据长度短,质量差。

- 比例
需要关注的是两个比例:
一个是subreads与polymerase reads数据量的比例,比例过低反映测序过程中的低质量的序列较多;
一个是zmw孔载入的比例,根据孔中载入的DNA片段数分为P0、P1和P2。P1合理比例在40%-60%之间。上样浓度异常会导致P0或P2比例过高,有效数据量减少。需要注意的是P2比例过低时,可能存在P2转P1的情况,测序结果包含较多的嵌合型reads。
 PacBio QC-quality zmw loading
PacBio QC-quality zmw loading一张芯片上有15万个孔,其中只有大概三分之一有一个测序复合物(聚合酶+测序引物+测序模版),另外三分之一是空的,剩下的三分之一是有>2个以上的测序复合物产生的数据再接下来的分析中是要去掉
组装
目前采用的组装策略:
PacBio-only de novo assembly :只使用 PacBio 产生的 long reads 进行拼接,在拼接之前要进行预处理,然后采用 Overlap-Layout-Consensus 算法进行拼接
Hybrid de novo assembly :结合 PacBio 的长reads 和 二代的短 reads
Gap filling :用二代的短reads(包括Pair-end和Mate-pair reads)拼接得到scaffod,然后用 PacBio 的长 reads 进行补洞
Scaffolding :用二代的短reads(包括Pair-end和Mate-pair reads)拼接得到 contigs / scaffod,用 PacBio 的长 reads 确定 contigs / scaffod 之间的位置关系

这四种组装策略并不是完全孤立的,在一个组装任务的不同阶段会用到不同的方法
不同的组装策略可以选用的工具:
-
PacBio-only
- HGAP:先进行reads的预组装(preassembly),然后用Celera® Assembler进行进一步组装,最后用 Quiver 进行校正
- Falcon:一个试验性的二倍体组装工具,已经在Gb级别大小的基因组上做了试验
- Canu:以Celera Assembler为基础,为三代单分子测序而开发出的分支工具
- Celera® Assembler:现在,Celera® Assembler 8.1 已经可以直接用于subreads的组装
-
Hybrid
- pacBioToCA:Celera® Assembler的一个error correction模块,最初是用来align short reads to PacBio reads 和 generate consensus sequences。随后,这些错误校正过的PacBio reads可以用Celera® Assembler进行组装
- ECTools:使用 unitigs (High quality contigs formed from unambiguous, unique overlaps of reads) 而非short reads进行校正
- SPAdes :SPAdes原本是进行短序列组装,在3.0版本后增加了对PacBio的混合组装的支持
- Cerulean :用ABySS构建de Bruijn graph,在图的bubbles位置利用PacBio的long reads解决bubbles带来的分支选择问题,从而延伸contigs
-
Gap Filling
PBJelly 2 :对已经组装过的基因组,用PacBio的long reads进行补洞

de novo assembly 算法
基因组的组装问题,实际上就是从序列得到的图中搜寻遍历路径的问题,有两种构建图的方法:
- overlap-layout-consensus (OLC)
- de Bruijn graph
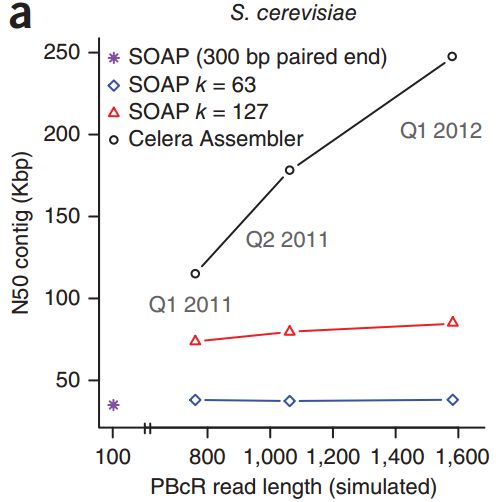
可以看到,随着reads长度的增加,基于OLC算法的组装工具组装出的contigs的长度几乎在线性增长,而基于de Bruijn图算法的组装效果并没有随着reads长度的增加而提高
三代单分子测序会产生较高的随机错误,平均正确率在82.1%-84.6%。这么高的错误率显然不能直接用于后续的分析,需要进行错误校正:
多测几个pass:由于测序序列是发夹结构,可以进行多轮的滚环测序,靠覆盖度来自我纠错,如果通量不是限制因素,那么PacBio是目前最准确的测序方式:错误率可以无限接近罕见突变的发生率(即无法分辨是测序错误还是罕见突变),不过这会极大缩短有效测序的插入序列的长度
用二代的短reads校正:2012年冷泉港实验室的Michael Schatz开发了一种纠错算法,用二代测序的短读长高精确数据对三代长读长数据进行纠错,这种称为”混合纠错拼接”(PBcR (PacBio corrected Reads) algorithm)
- Map short reads to long reads
- Trim long reads at coverage gaps
- Compute consensus for each long read

粉色长方形:单个PacBio RS reads;黑色竖线:测序错误;(a)由于测序错误碱基的存在使得两条reads就难确定是否在末端重叠;(b)高质量的短reads比对到存在错误的长reads;短reads中的黑色竖线表示 ‘mapping errors’ ,是长reads和短reads中测序错误的组合,此外双拷贝的重复序列的存在(灰色轮廓)导致在每一个拷贝中出现短reads的堆挤,为避免reads map到错误的重复区,仅保留最高比对值的短reads;(c)剩余的比对形成一致性序列(紫色长方形),长reads和短reads中共有的部分错误未能得到纠正;(d)overlap纠正后的长reads;(e) 最后的组装能够跨越重复区域。
校正过程中会将short reads未覆盖到的Gap进行裁剪,short reads在PacBio long reads上的覆盖情况:
这样做的其中一个考虑是去除adapter
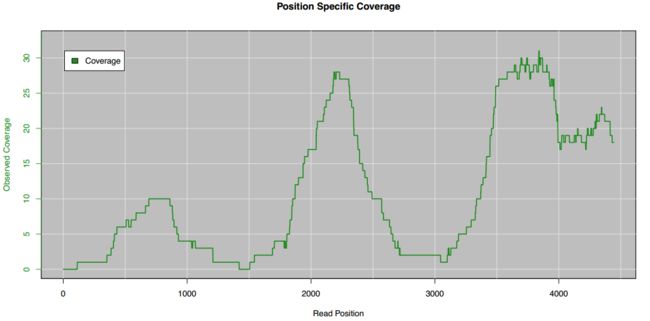
那么是什么原因导致了低覆盖度区域的产生的呢?
- Simple Repeats – Kmer Frequency Too High to Seed Overlaps
- GC Rich Regions – Known Illumina Bias
- Error Dense Regions – Difficult to compute overlaps with many errors
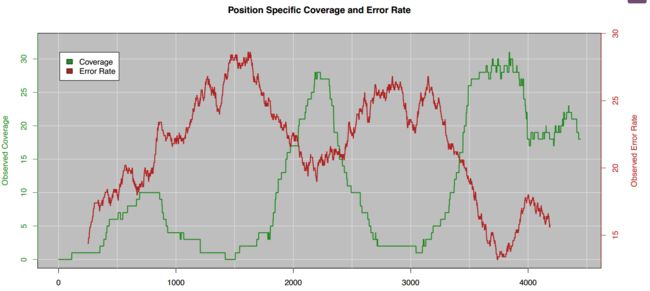 Position specific coverage and error rate
Position specific coverage and error rate
 3GS-assembly-error-correction-position-coverage-3.png
3GS-assembly-error-correction-position-coverage-3.png为了克服第三中情况导致的高测序错误率区域的低覆盖度,研究人员提出了用Unitigs进行校正的方法
 Pacbio error correction by Unitigs
Pacbio error correction by Unitigs
Nanopore数据分析
Base-calling
Base-calling做的就是从测序仪输出的电流信号波形图中将碱基解码 (decoding) 出来
第一步就是就是对波形图进行分段 (segmentation),即检测每个current shift的边界,这一步由ONT公司提供的 MinKNOW 完成,但是分段基于的假设是ssDNA分子匀速穿过nanopores,但是由于ssDNA穿过nanopore的速度很快,很容易产生一两个碱基的速度差异,这样就容易在decoding时造成insert和delete
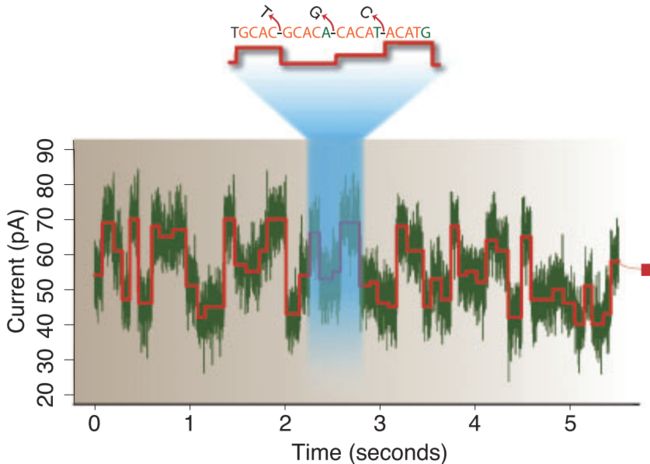
接着就基于current shift进行base calling,ONT公司提供的base caller为Metrichor,其底层算法基于HMM,将可能的k-tuple(由k个碱基组成的序列)作为隐藏状态,将current signals作为观测状态。ONT公司最新开发出的Metrichor用RNN取代了HMM,并将其整合到其开发出的新的生物信息数据分析平台EPI2ME中
随后,科研圈又开发出了开源的base calling工具,Nanocall 和 DeepNano。
- Nanocall类似于Metrichor,也是基于HMM。
- DeepNano 采用的是RNN(循环神经网络),又称为RNN base-caller,其输入为:mean, SD and duration of each segmented event ,其输出为各种碱基的概率分布。DeepNano在base calling准确率和计算速度上,都比ONT官方提供的Metrichor表现更好
DeepNano outperforms the Metrichor basecaller in terms of both accuracy (from 70 to 75%
sequence identityfor 1D read and from 85 to 87% for 2D reads) and computational speed
(190 s for a 2D read with Metrichor and 11 s with DeepNano)
ONT后来又在github上开源了一个RNN base-caller —— Nanonet
Data formats and handling
测序时,测序仪 MinION 连接上主机,安装在主机上的软件 MinKNOW 控制测序仪,对于每条reads,其 signal segmentation 结果(包括segment mean, variance and duration)以及测序过程中的 metadata 会被保存成FAST5格式的二进制文件(基于 HDF5标准 的变种)。
保存在FAST5文件中的原始数据会经过云端的Metrichor的处理,产生的解码的序列会被保存在另外的以.FAST5为后缀的HDF5文件中,包含一条template read和一条complement read或只有一条 2D read 。

MAP (MinION Access Programme) community 开发出的用于处理FAST5文件的工具,它们均能从FAST5文件中解析出FASTA/FASTQ文件,除此之外还有各自特色的质量统计功能:
Poretools: 输出quality plot,包括read-length histograms,yield-over-time
plots,和 squiggle plot (sequence of the segmented signals)NanoOK:评估三种类型的测序错误(substitutions, insertions and deletions),并绘制errors, coverage 和 k-mer 分布图
npReader:能够在测序进行过程中,进行实时评估,以GUI形式展示质量统计结果

参考资料:
(1) 生物技能树论坛:PacBio sequence error correction amd assemble via pacBioToCA
(2) 天津医科大学,伊现富《系统生物学-chapter2》
(3) Nanopore 第四代测序技术简介
(4) Magi A, Semeraro R, Mingrino A, et al. Nanopore sequencing data analysis: state of the art, applications and challenges.[J]. Briefings in Bioinformatics, 2017.
(5) 细节曝光!Oxford Nanopore真机还原,听听圈内人怎么说
(6) 三代测序--QC篇
(7) PacBio Training: Large Genome Assembly with PacBio Long Reads
(8) Koren S, Schatz M C, Walenz B P, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads[J]. Nature Biotechnology, 2012, 30(7):693-700.
(9) 冷泉港ppt:Hybrid De Novo Assembly of Eukaryo6c Genomes
(10) Leggett R M, Darren H, Mario C, et al. NanoOK: multi-reference alignment analysis of nanopore sequencing data, quality and error profiles[J]. Bioinformatics, 2016, 32(1):142-144.