Stratification of additional genomic features by TMB and Tcell–inflamed GEP
TMB和tcell炎症性GEP对其他基因组特征的分层
-
The patient groups defined by TMB and GEP status show notable differences in clinical response to pembrolizumab.
以TMB和GEP状态定义的患者组对pembrolizumab的临床反应有显著差异。
-
Inparticular,the two groups with only one positive biomarker indicative of potential for pembrolizumab response(TMBhi GEPlo or TMBlo GEPhi) have markedly lower response rates than the TMBhi GEPhi group,suggesting that mechanisms of resistance to pembrolizumab may exist that are specific to each respective group.
特别是,只有一个阳性生物标志物提示pembrolizumab潜在应答的两组(TMBhi GEPlo或TMBlo GEPhi)的应答率明显低于TMBhi GEPhi组,这表明对pembrolizumab的耐药机制可能存在,且可能针对每一组。
In order to identify potential mechanisms of resistance,we assessed molecular differences among tumors that belong to differentTMB-andTcell– inflamed GEP–defined groups through analyses in TCGA molecular database.
为了确定潜在的耐药机制,我们通过对TCGA分子数据库的分析,评估了不同tmb和tcell -炎性gep组肿瘤之间的分子差异。
First, we compared the correlation of genes in the transcriptome with GEP in TMBhi and in TMBlo tumors separately.
首先,我们分别比较了TMBhi和TMBlo肿瘤中转录组基因与GEP的相关性。
Both distributions of correlationsdivergedfromanormaldistribution because of a pattern of significant skewing toward positive correlations with the T cell– inflamed GEP, consistent with robust coregulation of gene expression markers of cell types present in a cytolytic TME.
这两种相关性的分布都偏离了正态分布,因为一种与T细胞发炎的GEP呈显著正相关的模式,这与溶细胞TME中细胞类型的基因表达标志物的强大协同调控一致。
However, there were no major differences in the correlations of individual genes with the T cell–inflamed GEP betweenTMBhi (TMB>100mutationsperexome) and TMBlo (TMB ≤ 100 mutations per exome tumors (r = 0.76;P < 1 × 10 −20) (Fig. 5B), suggesting a lack of qualitative difference in T cell inflammation markers as a function of tumor neoantigenicity.
然而,单个基因与T细胞炎症性GEP的相关性在tmbhi ((TMB>100突变/外显子组)和TMBlo (TMB≤100个突变每个外显体肿瘤)之间没有显著差异(r = 0.76;P < 1×10−20)(图5B),表明T细胞炎症标志物作为肿瘤新抗原性的功能缺乏定性差异。
[图片上传失败...(image-3537dd-1556593615660)]
Notably,muchsmallerdeviations from a normal distribution were observed in the negative range of correlations with GEP in both TMBhi andTMBlo tumors,suggesting the absence of major pan-cancer transcriptional signatures strongly associated with T cell exclusion.
值得注意的是,在TMBhi和tmblo肿瘤中,GEP与正态分布的负相关范围内的偏差要小得多,这表明没有与T细胞排斥密切相关的主要泛癌转录特征。
To understand the origin of the skewness toward positive correlations with the T cell– inflamed GEP, genes positively correlated with the T cell–inflamed GEP(r>0.15)were classified into two sets by using cutoffs defined by deviations from a normal distribution of the correlation with the T cell–inflamed GEP at 83% and 98% quantiles, respectively (Fig. 5C).
理解偏态向正相关性的起源与T细胞- GEP发炎,基因与T cell-inflamed GEP呈正相关(r > 0.15)分为两组通过达标由偏离正态分布的相关性与T cell-inflamed GEP 分别83%和98%分位数,(图5C)。
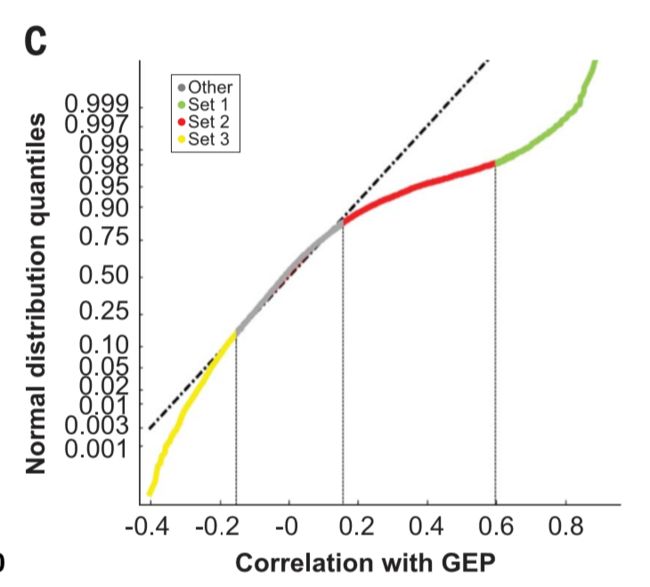 fig5C
fig5C
-
Set 1 comprisedgenesthathadaSpearmancorrelationr> 0.6 with the T cell–inflamed GEP (the lower bound for the correlation of individual genes inthe signature with the signature as awhole), whereas set 2 genes had correlations with GEP that ranged between 0.15 and 0.6.
Set 1与T细胞感染的GEP(签名中单个基因与整个签名相关性的下界)相关,而Set 2与GEP相关,范围在0.15到0.6之间。
-
Additionally,genes negatively correlated with the T cell– inflamed GEP and divergent from a normal distribution (r < −0.15 at 14% quantile) were grouped in set 3
此外,与T细胞炎症性GEP呈负相关并偏离正态分布(r < - 0.15, 14%分位数)的基因分组在set 3中.
As expected, a strong enrichment of genes relatedtoTcell–inflamed cytolytic processeswas observed in set 1 (table S5).
正如所料,在set 1中观察到与全细胞炎症溶细胞过程相关的基因大量富集(表S5)。
By contrast, set 2 showed enrichment in genes specific to other cell types in the TME, including vascular endothelium and myeloid infiltrate, but did not show enrichmentofgenesforT cell–inflamed cytolytic processes or tumorcell–intrinsic pathways.
相比之下,set 2显示了TME中其他细胞类型特异性基因的富集,包括血管内皮和髓样浸润,但没有显示丰富的细胞炎症溶细胞过程或肿瘤细胞固有通路。
Genes in set 1 and set 2 were further grouped as modules of gene coexpression by K-means clustering(K=10 forset2,andK=4 forset1).
通过K-means聚类(K=10 forset2, K=4 forset1),将set1和set2中的基因进一步分组为基因共表达模块。
Modules in set 1 did not show a strong association with TMB, consistent with the weak associations between TMB and the T cell–inflamed GEP described above.
set 1中的模块没有显示出与TMB的强相关性,这与上面描述的TMB与T细胞感染的GEP之间的弱相关性一致。
However, severalmodules in set 2 (table S6) displayed distinct patterns of correlation or anticorrelation with TMB.
然而,set 2(表S6)中的几个模块显示了与TMB不同的关联或抗腐蚀模式。
Annotation of the genes in the modules that were most strongly correlated and anti correlated with TMB (modules 4 and 5, respectively), revealed enrichment in biology related to cell proliferation(module4) and vasculature (module 5). These data suggest that distinct patterns of underlying biology can be identified by using TMB and the T cell– inflamed GEP to categorize tumors(Fig.5D).
注释的基因最强烈相关的模块和反与TMB (分别为模块4和5),揭示了富集在生物学相关细胞增殖(module4)和脉管系统(模块5)。这些数据表明,不同的潜在生物学模式可以使用TMB和T细胞——发炎GEP对肿瘤进行分类(Fig.5D)。
(正态偏离追寻加基因富集分析,找到了弱相关的因素。)

-
The association of the average expression of these gene modules(modules4 and 5)with TMB and T cell –inflamed GEP is represented in Fig. 5D in the up per l eft and lower right panels,respectively, by using the cytolytic module 1 from set 1 in the upper right panel as a reference.
联系的平均表达这些基因模块(modules4和5)TMB和T细胞发炎GEP表示在图5 d / 左和右下板,分别利用细胞溶解的模块1组1在右上角面板作为参考。
The group of genes in set 3 that were anticorrelated with the T cell–inflamed GEP (r < −0.15) was also investigated;
还研究了set 3中与T细胞炎症GEP负相关的基因组(r < - 0.15);
however, the biological annotation of the resulting coexpression moduleswaslessinformativethanthatforgenes positively correlated with the T cell–inflamed GEP.
然而,与与T细胞感染的GEP正相关的基因相比,由此产生的共表达调控的生物学注释所提供的信息更少。
However, some modules in this group were anticorrelated with TMB as well as with T cell –inflamed GEP.
然而,这一组中的一些模块与TMB以及T细胞炎症的GEP有负关系。
In particular, a module enriched in stromal and Wnt signaling elements was identified in tumors with both TMBlo and T cell –inflamed GEPlo (Fig. 5D, lower left panel).
特别是,在TMBlo和T细胞发炎的GEPlo肿瘤中发现了一个富含基质和Wnt信号元件的模块(图5D,左下面板)。
An additional analysis was performed by interrogating the entire transcriptome for genes associated with TMB in T cell–inflamed tumors, independently of the GEP-based clustering approach described above.
通过询问整个转录组,独立于上述基于gep的聚类方法,对T细胞炎症性肿瘤中与TMB相关的基因进行了额外的分析。
-
Similar to the analysis of modules, this analysis showed that genes that positively correlated with TMB were enriched for proliferation whereas those that were anticorrelated with TMB were related to vascular and stromal biology (table S7).
与模块分析相似,本分析表明,与TMB正相关的基因在增殖方面得到了富集,而与TMB抗负相关的基因则与血管和基质生物学相关(表S7)。
Consistent with these analyses,the distribution of previously identified signaturesof stromalbiology, proliferation, cytolytic activity, and Wnt signaling (13, 32–34) also showed similar patterns of association with TMB and theTcell–inflamed GEP(fig.S6).
与这些分析一致,先前识别的基质生物学、增殖、细胞水解活性和Wnt信号的分布(13,32 - 34)也显示了与TMB和tcell炎症的GEP相似的关联模式(图s6)。
However,in this analysis, we were not able to identify a gene expression signature of TMBhi that was as predictive as TMB itselff or response to pembrolizumab
然而,在这项分析中,我们不能确定TMBhi的基因表达特征,这与TMB本身或对pembrolizumab的反应一样具有预测性。
A complementary approach was used to identify genomic determinants of low cytolytic transcriptomic activity (absence of a T cell–inflamed GEP) in tumors with TMBhi as potential drivers of immune evasion in a mutagen-rich context.
一种互补的方法被用来确定在突变体丰富的情况下,TMBhi作为免疫逃避的潜在驱动因素的肿瘤中,低溶细胞转录组活性(没有T细胞发炎的GEP)的基因组决定因素。
As described above, the transcriptomic correlation oftheT cell–inflamed GEPinTMBhi tumors(Fig.5B) showed a distribution that skewed toward positive correlation with GEP, suggesting the absence of a robust transcriptome signal in tumors with TMBhi and GEPlo.
如上所述,t细胞炎症GEP和TMBhi肿瘤的转录组相关性(图5b)呈与GEP呈正相关的分布,提示TMBhi和GEPlo肿瘤中缺乏一个强的转录组信号。
Therefore, DNA alterations in TCGA were explored to reveal potential negative associations of somatic mutations with GEP by using a previously reported approach(13)butfocusingspecificallyontumors withTMBhi.
因此,利用先前报道的方法(13)研究TCGA的DNA变化,以TMBhi为重点,揭示了体细胞突变与GEP之间潜在的负相关关系。
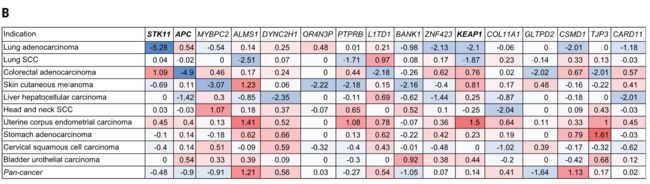
Among known cancer drivers serinethreonine kinase 11 (STK11) [also known as liver kinase B1 (LKB1)] mutation in lung adenocarcinoma,Kelch-likeECH-associated protein1(KEAP1) mutation in lung adenocarcinoma and lung squamous cell carcinoma, and adenomatous polyposis coli (APC) mutation in colorectal cancer showed highly significant negative associations with the T cell–inflamed GEP (Fig. 6). Notably,
已知的癌症驱动serinethreonine激酶11 (STK11)(也称为肝激酶B1 (LKB1)]在肺腺癌突变,Kelch-likeECH-associated protein1 (KEAP1)突变在肺腺癌和肺鳞状细胞癌,腺瘤息肉杆菌(APC)的突变结直肠癌显示高度显著的负相关T cell-inflamed GEP(图6)。
Notably,none of these associations passed the nominal significance level(P<0.01)in the pan-cancer analysis, suggesting a potential cancer type–specific role for these somatic alterations.
值得注意的是,,这些关联在泛癌分析中均未超过名义显著性水平(P<0.01),提示这些躯体改变可能与癌症类型特异性有关。
Other genes demonstrating negative associations with the T cell– inflamed GEP were either of low frequency or were not known cancer drivers (Fig. 6B).
其他与T细胞炎症性GEP呈负相关的基因要么是低频率的,要么是未知的癌症驱动因素(图6B)。
(深入研究基因相关关系,与与GEP正/负相关的基因)
Discussion
-
Several studies have shown that either TMBhi or cytolyticelementsoftheTMEareassociatedwith clinical response to checkpoint blockade immunotherapy in some tumor types (4–9, 11–13, 15).
几项研究表明,TMBhi或溶细胞因子与某些肿瘤类型(4 - 9,11 - 13,15)对检查点阻断免疫治疗的临床反应有关。
However, the relationship between these two central aspects of tumor immunobiology and their combined associationwithclinical response to checkpoint blockade immunotherapy has not been well-studied across multiple cancer types.
然而,肿瘤免疫生物学的这两个核心方面之间的关系,以及它们与检查点阻断免疫治疗的临床反应的联合关系,在多种癌症类型中尚未得到很好的研究。
-
Here, we show that TMB and a T cell–inflamed GEP are tissue-agnostic measures of distinct aspects oftumor immunobiology and independently predict response to anti–PD-1 therapy in multiple tumors.
在这里,我们证明TMB和T细胞炎症的GEP是肿瘤免疫生物学不同方面的组织不可知的测量方法,并独立预测抗pd -1治疗在多种肿瘤中的反应。
In particular, limited clinical responses to pembrolizumab occurred in patients with low levels of both TMB and T cell– inflamed GEP, whereas the greatest response rates were seen in patients with high levels of bothbiomarkers.
特别是,对pembrolizumab的临床反应有限发生在TMB和T细胞炎症性GEP水平较低的患者中,而对这两种生物标志物水平较高的患者的反应率最高。
-
Similarly,improvedresponses were seen in patients who had high levels of both PD-L1 IHC expression and TMB, reflective of the relationship of PD-L1and GEP to a Tcell– inflamed TME.
同样,PD-L1 IHC和TMB表达水平高的患者反应也有所改善,这反映了PD-L1和GEP与Tcell炎症性TME的关系。
-
These observations suggest that using inflammatory biomarkers such as the T cell –inflamed GEP or PD-L1 jointly with TMB may helpto identifypatientswho are responsive toanti–PD-1 therapies.
这些观察表明,使用炎症生物标志物,如T细胞发炎的GEP或PD-L1与TMB联合使用,可能有助于识别对抗pd -1治疗有反应的患者。
AdditionalIHCassayshave been developed thatmeasureproteinmarkersof a cytolytic T cell environment, and evaluating their performance characteristics in conjunction with TMB in future studies may be useful (14, 35).
此外,已经开发出一种方法来测量溶细胞T细胞环境的蛋白标记,并在未来的研究中结合TMB来评估它们的性能特征,这可能是有用的(14,35)
More broadly, our study demonstrates the orthogonal relationship between universal measures oftumor antigenicityandtumorinfiltrationthat can occur by activated T cells (14, 36–38).
更广泛地说,我们的研究表明,普遍的肿瘤抗原检测方法与激活的T细胞可能发生的肿瘤浸润之间存在正交关系(14,36 - 38)。
Although these are upstream and downstream components, respectively, of a robust antitumor T cell response, there is sufficient intervening biology such that biomarkers for each process can provide complementary information.
虽然这些分别是抗肿瘤T细胞反应的上游和下游成分,但有足够的生物干预,使得每个过程的生物标志物可以提供补充信息。
As an increasing number ofPD-1– and PD-L1– based combination regimens show clinical benefit, it will become challenging to determine the relative utility of each regimen for an individual patient.
随着越来越多基于pd -1和PD-L1的联合方案显示出临床效益,确定每种方案对单个患者的相对效用将变得具有挑战性。
A refined setof biomarker toolsthatcan stratify underlying patterns of tumor immunobiology may enable rational and biology-driven personalization of these various treatment regimens mens, such as selection of patients with tumors typically less responsive to immunotherapy.
一套能够对肿瘤免疫生物学潜在模式进行分层的生物标志物工具,可能会使这些不同治疗方案的患者在生物学驱动下实现合理的个性化,比如选择对免疫治疗通常反应较慢的肿瘤患者。
Our datademonstratethatTMBandaTcell–inflamed GEP can be used to categorize tumors into discrete subgroups that exhibit distinct patterns of potentially targetable biology to enhance clinical response.
我们的数据策略是,带状细胞炎症性GEP可用于将肿瘤划分为不同的亚组,这些亚组具有不同的潜在靶向生物学模式,以增强临床反应。
These patterns include tumor type– agnostic signatures of proliferative, vascular, myeloid, and stromal biology, as well as tumor type–specificdysregulationoftumorcell–intrinsic signaling pathways.
这些模式包括肿瘤类型不可知的特征增殖,血管,骨髓和基质生物学,以及肿瘤类型特异性的肿瘤细胞固有信号通路失调。
Although the utility of TMB, T cell –inflamed GEP, and PD-L1, as well as other emerging tumor-agnostic biomarkers, will need to be prospectively validated for use in predicting response to various immunotherapy regimens, including combination therapies, the findings reported here suggest a rationale for further exploring the utility of these biomarkers as guides for precision cancer immunotherapy.
尽管TMB的效用,T细胞发炎GEP和PD-L1,以及其他新兴tumor-agnostic生物标记,需要前瞻性验证用于预测应对各种免疫治疗方案,包括联合疗法,研究结果报道在这里建议理由进一步探索这些生物标记物的效用作为精密癌症免疫治疗的指南。
(利用这种相关关系,预测免疫治疗的有效性,提供临床指导方案,希望在以后可以T细胞发炎GEP和PD-L1的分子信息获得肿瘤的特征,肿瘤类型,特性性肿瘤固有的信号通路等信息,指导临床治疗)
Materials and methods Clinical tumor samples
-
Associations of TMB and the T cell–inflamed GEP with BOR and PFS were evaluated by using tumor samples from subgroups of patients treated with pembrolizumab in clinical trials who had WES data available.
通过使用临床试验中接受pembrolizumab治疗的患者亚组的肿瘤样本,对TMB和T细胞炎症性GEP与BOR和PFS的关系进行了评估,这些患者拥有WES数据。
These included a discovery cohortofpatientswithHNSCC(KEYNOTE-012 B1),a pan-tumor validation cohort(KEYNOTE012/028), and single-indication cohorts of patients with HNSCC(KEYNOTE-012B1+B2) and melanoma(KN001and006).
其中包括一项与HNSCC(KEYNOTE-012B1)的患者的发现(cohortofpatientswithHNSCC),一个泛肿瘤验证队列(KEYNOTE012/028),以及与HNSCC(KEYNOTE-012B1+B2)和黑色素瘤(kn001和006)患者的单指征队列。
-
The discovery cohort included 34 of 297 total enrolled patients with PDL1–selected (≥1%, modified proportion score or interface pattern, QualTek IHC) (39) HNSCC (B1 cohort).
发现队列包括297例入选患者中的34例(PDL1-selected≥1%,modified proportion score or interface pattern, QualTek IHC) (39) HNSCC (B1队列)。
-
The pan-tumor cohort comprised patients with PD-L1–positive (≥1%, modified proportion score or interface pattern, QualTek IHC) (39) advanced solid tumors pooled from two multicohort trials, including 39 of 297 total enrolled patients in KEYNOTE-012(cohortsA,C,and D:triple-negative breast cancer, urothelial cancer, and gastric cancer,respectively)and 80 of 450 total enrolled patients in KEYNOTE-028(17of20cohorts with anal, biliary, carcinoid, cervical, colorectal,endometrial,esophageal,estrogen receptor–positive human epidermal growth factor receptor-2–negative breast, pancreatic, salivary gland, prostate, small cell lung, thyroid, and vulvar cancers and neuroendocrine tumors, mesothelioma, and leiomyosarcoma).
pan-tumor队列由PD-L1-positive患者(分数比例≥1%,修改或接口模式,QualTek包含IHC)(39)高级实体肿瘤合并两个multicohort试验,包括39 297总登记病人的主题- 012 (cohortsA, C和D:三阴性乳腺癌,移行细胞癌,胃癌,分别)450年和80年总登记病人主题- 028 (17 of20cohorts肛门、胆道良性肿瘤,宫颈,结直肠、子宫内膜、食管、雌激素受体阳性的人表皮生长因子受体2阴性的乳腺、胰腺、唾液腺、前列腺、小细胞肺癌、甲状腺、外阴癌、神经内分泌肿瘤、间皮瘤和平滑肌肉瘤)。
Single-indication cohorts included 107 HNSCC patients from the KEYNOTE-012PD-L1– positive(≥1%, modified proportion scoreor interface pattern, QualTek IHC) (39) B1 ( n = 34) and PD-L1–unselectedB2(n=73)cohorts(40,41)and patients with advanced melanoma from the pembrolizumab arms of the KEYNOTE-001 (n = 30 of 668 total enrolled patients) and KEYNOTE006 (n=59 of 834 total enrolled patients)studies (26, 42).
Single-indication组包括107 HNSCC病人的 KEYNOTE- 012 - pd - l1 -积极(≥1%,修改比例scoreor接口模式,QualTek包含IHC) (39) B1 (n = 34)和PD-L1-unselectedB2 (n = 73)组(40、41)和晚期黑色素瘤患者pembrolizumab武器的 KEYNOTE- 001 (n = 30 668总登记的病人)和KEYNOTE006 (n = 59 834总登记病人)的研究(26日42)。
Tissue specimens were obtained with the approval ofthe institutional review boards, and patients provided informed consent [clinical trial registration: KEYNOTE-012 (NCT01848834);KEYNOTE-028 (NCT02054806);KEYNOTE-001 (NCT01295827);KEYNOTE-006 (NCT01866319)].
组织标本获得机构审查委员会批准,患者提供知情同意[临床试验注册:KEYNOTE-012 (NCT01848834);KEYNOTE-028 (NCT02054806);KEYNOTE-001 (NCT01295827);KEYNOTE-006 (NCT01866319)].
Clinical end points
-
BOR was assessed in the discovery HNSCC, pantumor, and HNSCC cohorts by central radiology reviewandinthemelanomacohortbyintegrated radiology and oncologist assessment.
BOR在发现HNSCC、pantumor和HNSCC组中通过中央放射学评论和综合放射学和肿瘤学评估进行评估。
-
For BOR, a responder was defined as a patient with a partial response(PR)orcompleteresponse(CR),andPFS wasdefinedasthetimefromthestartoftreatment to documented evidence of progressive disease or death.
对于BOR,应答者被定义为部分应答(PR)或完全应答(CR)的患者,pfs被定义为从开始治疗到有记录的渐进性疾病或死亡证据的时间。
-
BOR and PFS were both assessed in the all patients-as-treated populations,defined as those who had received ≥1dose of study drug,in each cohort
在每个队列中,所有接受治疗的患者(定义为接受≥1剂量研究药物的人群)均进行BOR和PFS评估
Processing of tissue samples
-
DNA sequencing (WES) and RNA analysis (gene expression profiling) were performed by using FFPE sections of pretreatment tumor samples fromtheabove-listedstudies.
采用上述研究中预处理肿瘤标本的FFPE切片进行DNA测序(WES)和RNA分析(基因表达谱)。
-
WESwasperformed onbothgermlineandtumorsamples,andgene expression profiling was performed on tumor samples.
我们对细胞和肿瘤样本进行了检测,并对肿瘤样本进行了基因表达谱分析。
-
With a fresh scalpel, the tissue was either macrodissected from the marked tumor area (tissue containing <20% tumor) or scraped fromtheentiresectionandtransferredtoa1.5-ml tube containing 200 ml of 100% ethanol
用新鲜的手术刀,从标记的肿瘤区域(肿瘤组织小于20%)大范围切除组织,或从整个切片上刮取组织,转移到一个1.5 ml的试管中,试管中含有200 ml的100%乙醇
Gene expression (RNA) profiling: NanoString methodology
-
The previously described T cell–inflamed GEP was derived by using a stepwise derivation process of discovery, validation, and refinement of candidate genesets acrossawidevariety of solid tumors(15).
先前描述的T细胞炎症性GEP是通过发现、验证和纯化多种实体肿瘤候选基因集的逐步衍生过程而得到的(15)。
-
The GEP was composed of 18 inflammatory genes related to antigen presentation, chemokine expression, cytolytic activity, and adaptive immune resistance, including CCL5, CD27, CD274 (PD-L1), CD276 (B7-H3), CD8A, CMKLR1, CXCL9, CXCR6, HLA-DQA1, HLA-DRB1, HLA-E, IDO1, LAG3, NKG7, PDCD1LG2 (PDL2), PSMB10,STAT1,andTIGIT.
GEP由CCL5、CD27、CD274 (PD-L1)、CD276 (B7-H3)、CD8A、CMKLR1、CXCL9、CXCR6、HLA-DQA1、HLA-DRB1、HLA-E、IDO1、LAG3、NKG7、PDCD1LG2 (PDL2)、PSMB10、STAT1、tigit等18个与抗原表达、趋化因子表达、细胞水解活性、适应性免疫耐受相关的炎症基因组成。
-
For GEP analysis,total RNA was isolated from 5-mm-thick FFPE sections of tumor tissue fixed on positively charged slides (Ambion Recover All total nucleic acid isolationkit for FFPE;catalog no.AM1975) at ALMAC, United Kingdom.
GEP分析从固定于带正电荷载玻片上的肿瘤组织5 mm厚的FFPE切片中提取总RNA (Ambion回收所有用于FFPE的总核酸分离试剂盒;
-
Total RNA concentrations were measured using the NanoDrop ND1000 (Thermo Fisher Scientific) in 1.5 ml of test sample.
使用NanoDrop ND1000 (Thermo Fisher Scientific)在1.5 ml的测试样品中测定总RNA浓度。
-
Gene expression analysis was conducted on the NanoString nCounter gene expression platform (NanoString Technologies, Seattle, WA) as described previously (15).
如前所述,在NanoString nCounter基因表达平台(NanoString Technologies, Seattle,WA)上进行基因表达分析(15)。
-
Per sample, 50 ng of total RNA was mixed in a final volume of 5 to 7 ml with a 3 ′-biotinylated capture probe and 5′-reporter probe tagged with a fluorescent barcode, from the desired custom gene expression codeset (HUIMR680_V2_C2406+PLS_SPIKE80_C2765 for Batch 1 and HUIMR800_C3176 for Batch 2), containing probes designed to function as positive and negative hybridization controls.
每个样本,50 ng的总RNA混合在最后一卷5到7毫升3′生物素化的捕获探针和5′记者探针荧光条码标记,从基因表达所需的自定义代码集(HUIMR680_V2_C2406 + PLS_SPIKE80_C2765批次1和HUIMR800_C3176批2),包含探测器设计定位为积极的和消极的杂化控制。
Probes and target transcripts were hybridized overnight at 65°C for 14 to 18 hours as permanufacturers’recommendations.
探针和目标转录本在65°C条件下杂交14 - 18小时,这是制造商的建议。
Hybridized samples were run on the NanoString nCounter preparationstationbyusingahigh-sensitivityprotocol where excess capture and reporter probes wereremovedandtranscript-specificternarycomplexes were immobilized on a streptavidin-coated cartridge.
杂交样品在纳米字符串非计数器制备站进行,使用高灵敏度的协议,其中过量的捕获和报告探针是由转录特异性的复合物固定在一个链霉亲和素涂层墨盒。
The cartridge samples were scanned at maximum resolution by using the nCounter digital analyzer.
使用nCounter数字分析仪以最大分辨率扫描墨盒样品。
GEP scores were calculated as a weighted sum of normalized expression values for the 18 genes.
GEP评分计算为18个基因归一化表达值的加权和。
Quality control of the gene expression data followed an approach similar to that of the NanoString clinical-grade assay, with theuseofjointcriteriathatassessedtherelationships between housekeeping genes and the negative control probes plus a weighted score evaluating the GEP gene counts versus background subtracted counts.
基因表达数据的质量控制采用了一种类似于纳米字符串临床级检测的方法,使用联合标准分析了内家基因和阴性对照探针之间的关系,并使用加权评分来评估GEP基因计数与背景减除计数之间的关系。
For housekeeping normalization, raw counts for the individual genes were log10 transformed and then normalized by subtracting the arithmetric mean of the log10 counts for a set of 11 housekeeping genes.
对于管家化,对单个基因的原始计数进行log10转换,然后通过减去一组11个管家化基因的log10计数的算术平均值进行归一化。
WES pipeline
Somatic single-nucleotide variant (SNV) calling
-
Whole-exome sequence reads were aligned to reference human genome GRCh37 by using bwa mem(43)followedbypreprocessingstepsincluding duplicate marking, indel realignment, and baserecalibrationwithPicard(v1.114)andGATK (Genome Analysis Toolkit, v2) (44) to generate analysis-ready BAM files.
通过使用bwa mem(43)和预处理步骤(包括重复标记、indel重新排列和使用picard (v1.114)和gatk(基因组分析工具包v2)(44)对整个外显子组序列进行比对,以参考人类基因组GRCh37。
MuTect-called SNVs present in the Single Nucleotide Polymorphism Database (dbSNP, v141) (46) but not in the CatalogueofSomaticMutationsinCancer (COSMIC, v68)(47)werefilteredout.TheSNVswithmutant reads of <4 in tumor samples were also eliminated.
MuTect-called SNVs出现在单核苷酸多态性数据库(dbSNP, v141)(46)中,但没有出现在体细胞突变目录sincancer (COSMIC, v68)(47)中,在肿瘤样本中,突变读数<4的sns也被剔除。
TMB for a subject was defined as the sum of somatic nonsynonymous SNVs thatpassed all the filters described
受试者的TMB被定义为通过所有描述的过滤器的躯体非同义snv之和
HLA class I typing
-
HLA-I major loci,A,B and C, weretyped atfourdigit resolution by using OptiType (v1.0) (48).
HLA-I主要位点A、B和C使用OptiType (v1.0)(48)以四位数分辨率输入。
-
For output typed alleles not found in the NetMHC(v3.4)(49)inputlist,thecorresponding supertype was identified for each allele (50, 51) and the supertype-representativeallele was used for NetMHC.
对于NetMHC(v3.4)(49)inputlist中没有发现的输出型等位基因,为每个等位基因(50,51)确定相应的超型,并将supertype- representative等位基因用于NetMHC。
SNV annotation and neoantigen detection
SNV注释和新抗原检测
-
Somatic mutations were annotated with VEP (Variant Effect Predictor) (52), and nonsynony mousmutations in protein codin gregions were counted for TMB.
用VEP(变异效应预测因子)标记体细胞突变(52),计算TMB中蛋白codin gregions中的非同步突变。
-
All possible 9-mer peptide sequences with mutated amino acid inside for each nonsynonymous mutation locus were extracted and binding affinities for patient HLA-AandHLAB alleles were computed by using NetMHC (v3.4).
提取每个非同义突变位点所有可能含有突变氨基酸的9-mer肽段序列,使用NetMHC计算患者HLA-AandHLAB等位基因的结合亲和力(v3.4)。
The 9-mer peptide with the highest binding affinity with the HLAalleles from a nonsynonymous mutation locus was selected as the representative antigen for the mutation.
选择非同义突变位点与hla等位基因结合亲和力最高的9-mer肽段作为该突变的代表性抗原。
Representative antigens with HLA-A or -B binding affinity of <50 nM were considered neoantigens.
具有代表性的HLA-A或-B结合亲和力<50 nM的抗原被认为是新抗原。
Microsatellite instability (MSI) calling
-
MSI phenotype was detected by applying mSINGS on WES data from tumor samples (22).
将mSINGS应用于肿瘤样本的WES数据,检测MSI表型(22)。
-
The stability of each mononucleotide microsatellite locus was evaluated, and the proportion of unstable microsatellite loci was determined as the MSI score.
对每个单核苷酸微卫星位点的稳定性进行评价,确定不稳定微卫星位点的比例作为MSI评分。
Samples with an MSI score of more than 20% were classified as MSI-high (MSI-H) positive.
MSI评分超过20%的样本为MSI-high (MSI- h)阳性。
-
MSIwasconfirmedbyPCRbyusingthe Promega MSI analysis system, version 1.2.
通过使用Promega MSI分析系统1.2版,msiwas得到了确认。
Mutation signature analysis
突变特征分析
-
Mutational signature analysis was performed by using the deconstructSigs package (v1.6.0) in R thatselectsthecombinationofknownmutational signaturesthatcanaccountfortheobservedmutational profile in each sample (53).
使用R中的解构tsigs包(v1.6.0)进行突变特征分析,该包选择能够解释每个样本中观察到的突变剖面的已知突变特征的组合(53)。
-
Exome regions were defined by Agilent Sureselect V5 target region.
由安捷伦Sureselect V5靶区定义外显子区。
-
Only somatic mutations in exome regions were considered, and trinucleotide counts were normalized by the number of times each trinucleotide context was observed in the exome region.
只考虑外显体区域的体细胞突变,并根据外显体区域中观察到的每个三核苷酸上下文的次数对三核苷酸计数进行标准化。
-
Mutational signatures as defined by Alexandrov et al.(54) and named as signatures.
由Alexandrov等人(54)定义并命名为签名的突变签名。
-
nature 2013 were the target signature set to be screened.
《自然》杂志(nature)将于2013年对目标签名进行筛选。
-
The relationships of these various mutational signatures, including specific nucleotide changes, DNA repair, smoking, neoantigen, TP53, and APOBEC, with BOR and PFS were evaluated in patient samples in the pan-tumor cohort.
这些不同的突变特征,包括特异性核苷酸变化、DNA修复、吸烟、新抗原、TP53和APOBEC,与BOR和PFS的关系在泛肿瘤队列患者样本中进行了评估。
Allele-specific copy number and purity estimation
等位基因特异性拷贝数和纯度估计
-
VarScan2 (55) output copy number ratio and SNP were input to Sequenza (56) to provide a maximum a posteriori estimation for cellularity and segmented allele-specific copy number for each sample.
VarScan2(55)输出拷贝数比和SNP被输入到Sequenza(56),为每个样本的细胞数量和分段等位基因特异性拷贝数提供最大的后向估计。
Clonality
-
Foreachsample,MuTect-calledsomaticSNVswith variant allele frequency information, combined with Sequenza output allele-specific copy number andcellularityestimation,wereinputtoPyClone to estimate cellular prevalence for all somatic SNVs.
本研究以含有变异等位基因频率信息的多克隆体snv为样本,结合Sequenza输出的等位基因特异性拷贝数和细胞密度估计,输入克隆体来估计所有体细胞snv的细胞流行率。
-
Mutational clonality was also inferred through the clustering process of PyClone
通过PyClone的聚类过程推断出突变克隆的克隆性
PD-L1 expression
-
PD-L1 expression levels were evaluated in pretreatment samples by IHC staining by using the PD-L1 IHC 22C3 pharmDx kit (Agilent Technologies)inthepan-tumor and HNSCC cohorts(39);
使用PD-L1 IHC 22C3药物dx试剂盒(Agilent Technologies)对肿瘤和HNSCC患者进行预处理,采用免疫组化染色法检测PD-L1的表达水平(39);
-
expression levels were reported as the CPS, definedasthenumberofPD-L1–positivecells(tumor cells,lymphocytes,macrophages)dividedbythe total number of tumor cells × 100.
表达水平以CPS表示,定义为肿瘤细胞(肿瘤细胞、淋巴细胞、巨噬细胞)被肿瘤细胞总数×100分。
-
CPS was previously reported as a percentage and is now reportedas an equivalentunitlessmeasure.
CPS以前被报道为一个百分比,现在被报道为一个等价的无单位度量。
This assay differs from the one used to determine PDL1 positivity (≥1%, modified proportion score or interface pattern, QualTek IHC) for enrollment eligibility as described above for the pan-tumor and HNSCC clinical cohorts (58).
本试验不同于用于确定PDL1阳性(≥1%,修改的比例分数或接口模式,QualTek IHC)的入选资格,如上所述的泛肿瘤和HNSCC临床队列(58)。
-
For the melanoma cohort, PD-L1 levels were assessed by IHC byusing the MEL score,andpositivitywasdefined as ascore of ≥2membranous PD-L1 staining in at least 1% of tumor and tumor immune cells (59)
对于黑色素瘤队列,使用MEL评分通过IHC评估PD-L1水平,阳性定义为至少1%的肿瘤和肿瘤免疫细胞中≥2膜性PD-L1染色(59)。
TCGA molecular data
Geneexpressiondatafor9963tumorsandsomatic alterations data for 6384 tumors were obtained through TCGA portal (16) as of September 2015
截至2015年9月,通过TCGA门户网站(16)获得6384例肿瘤的996363例肿瘤的基因表达数据和体细胞改变数据
Statistical methods
-
The retrospective, statistical analysis of clinical samples in this study was prespecified and performed in a blinded fashion, with genomic end points generated without access to clinical outcomes.
本研究中对临床样本的回顾性统计分析是预先指定的,以盲法进行,基因组终点的产生不涉及临床结果。
-
Associations with BOR were tested by using logistic regression, and associations with PFS were examined by using Cox proportional hazards models.
用logistic回归检验与BOR的相关性,用Cox比例危险模型检验与PFS的相关性。
-
All models (logistic regression and Cox models) were adjusted for baseline Eastern Cooperative Oncology Group (ECOG) score performance.
所有模型(logistic回归和Cox模型)均根据东部肿瘤合作组(ECOG)基线评分表现进行调整。
-
One-sided nominal P values were reported.
单边检测。
-
Associations between continuous variables were assessed by using Spearman correlation, and associations between continuous variables and binary variables (e.g., BOR) were further assessed by using AUROC and rank sum P values.
连续变量之间的相关性采用Spearman相关进行评估,连续变量与二元变量(如BOR)之间的相关性进一步采用AUROC和秩和P值进行评估。
-
Statistical analyses and visualizations wereperformedwithMatlabR2010orwithR3.4.1.
采用matlabr2010或withr3.4.1进行统计分析和可视化。
-
TMB cutoffs for the pan-tumor and singleindication clinical cohorts were the Youden Index values derived in AUROC analysis.
全肿瘤和单指征患者的TMB切断率是由AUROC分析得出的约登指数。
-
An additional,exploratory,pan-tumor TMB threshold was derived by using TMB and GEP data across each cohort, similar to a previously described method (20
另外一个探索性的泛肿瘤TMB阈值是通过使用每个队列的TMB和GEP数据推导出来的,类似于前面描述的方法(20)