
跟着Seurat团队学数学,从KNN到SNN到MNN到WNN,scRNA+时代与单细胞数据的统一场论。注意这里的NN不是 Neural Network(神经网络)而是Nearest Neighbor(最邻近)。NN的意思主要是在判定哪些细胞是一类的时候,用到的度量方式:谁们离得近就是一类。本文提出的WNN是在之前SNN( Shared Nearest Neighbor,共享邻近)和MNN(mutual nearest neighbor,互邻近)的基础上开发出来整合不同模态(如表面蛋白与转录组)的方法。本文翻译自Integrated analysis of multimodal single-cell data,个人水平有限,谬误之处在所难免,若蒙读者诸君不吝斧正,将感激不尽。
摘要
同时测量单个细胞的多模态数据,被称为多模态分析(multimodal analysis),代表了单细胞基因组学一个令人兴奋的前沿方向,同时也需要新的计算方法来基于多种数据类型描述细胞状态。在这里,我们介绍了“加权最近邻(weighted-nearest neighbor,WNN)”分析:一个无监督的框架来学习每个细胞中每种数据类型的相对效用,使多种模式的整合分析成为可能。将我们的程序应用于包含几十万个人类白细胞的CITE-seq数据集以及228个抗体panel,以构建一个循环免疫系统的多模态参考图谱。我们证明,整合分析大大提高了我们描述细胞状态的能力,并验证了以前未报道的淋巴亚群的存在。此外,我们还演示了如何利用这一参考来快速绘制新数据集,并解释免疫接种和COVID-19的免疫反应。我们的方法(WNN)代表了一种广泛适用的策略来分析单细胞多模态数据集,包括RNA和染色质状态的配对测量,并超越转录组来寻找细胞身份的统一和多模态定义。
安装说明、文档、教程和CITE-seq数据集可以在http://www.satijalab.org/seurat获得
介绍
对人类免疫系统中丰富多样的细胞类型进行分类和表征的潜力,为单细胞基因组学提供了一个强大的机会,但也显示出当前方法的局限性。虽然已建立的技术,如单细胞RNA-seq (scRNA-seq)能够发现异质组织中的新细胞类型和状态,但单靠转录组学常常无法分离分子上相似但功能上不同的免疫细胞类型。尽管T细胞具有巨大的功能多样性,但不同的T细胞群,如效应细胞、调节细胞、细胞内固定细胞和黏膜相关不变T细胞(MAIT),即使使用最敏感和最尖端的技术,通常也不能仅用scRNA-seq有效地分离它们。这反映了由T细胞中极少的RNA含量和高RNase表达所驱动的技术局限,也妨碍了scRNA-seq数据的质量。更广泛地说,这显示了仅基于转录组定义细胞状态所面临的困难,因为尽管在其他模式中可以识别,但细胞异质性的重要来源可能与转录组特征并不直接的密切相关。
多模态单细胞技术,在同一细胞中同时描述多种数据类型,代表了细胞状态发现和表征的新前沿。例如,我们最近引入了CITE-seq,它利用寡核苷酸偶联抗体,通过测序抗体衍生标签(antibody-derived tags ,ADTs),同时量化单细胞内RNA和表面蛋白的丰度。此外,随着技术进步,现在可以在染色质可及性(ATAC)、DNA甲基化、核小体占位(nucleosome occupancy )或空间定位的同时对转录组进行分析。这些方法都提供了一个令人兴奋的解决方案,以克服scRNA-seq固有的局限性,并探索多种细胞模式如何影响细胞状态和功能。
多模态单细胞技术的成熟就需要开发新的计算方法来集成不同数据类型的信息。例如,虽然CITE-seq数据集可以通过首先根据基因表达值聚类来分析,然后探索其免疫表型,而多模态计算工作流将基于这两种模式定义细胞状态。重要的是,这些策略必须是稳健的,能够应对每个模态在数据质量和信息内容潜在的差异。在某些情况下,稳健的蛋白质定量可能是最有用来分类的价值,特别是在一个大的和精心设计的抗体panel前提下。在其他情况下(如重要的细胞类型标记丢失或以前不知道时),细胞转录组的无监督聚类是比较有价值的。每一模态的不同信息内容,甚至在同一数据集中跨细胞的测量,提出了多模态数据集的分析和集成的迫切挑战。
在这里,我们引入了“加权最近邻”(weighted-nearest neighbor,WNN)方法,这是一个分析框架,用于集成细胞内测量的多种数据类型,并获得细胞状态的联合定义。我们的方法是基于非监督策略来学习细胞特定模态的“权重”,它反映每个模态的信息内容,并确定其在下游分析中的相对重要性。我们证明,WNN分析大大提高了我们定义多种生物数据类型中的细胞状态的能力。我们利用这种方法,基于包含211,000人外周血单核细胞(PBMC)的CITE-seq数据集生成多模式“图谱”,具有可扩展228个抗体的大细胞表面蛋白标记panel。利用这个数据集来识别和验证人类淋巴细胞中的异质细胞状态,并探索人类免疫系统对疫苗接种和SARS-CoV-2感染的反应。WNN在我们的开源R工具包Seurat的更新版本4中实现,代表了对单细胞数据进行综合多模态分析的广泛适用的策略。
结果
量化每个细胞中每个模式的相对效用
我们试图设计一个健壮的分析工作流程,以集成在同一细胞内收集的多个测量数据类型。要应用于一系列的生物模式和数据类型,我们的策略必须成功地满足以下标准。
- 首先,工作流必须是健壮的,以适应不同模式之间潜在的巨大数据质量差异。
- 第二,综合多模态分析应该能够实现多个下游分析任务,包括可视化、聚类和细胞轨迹的识别。
- 最后,也是最重要的是,与单独进行每种模式的独立分析相比,对多种模式的同时分析可以提高发现和表征细胞状态的能力。
这些挑战突出了使用灵活的框架来处理不同数据集的重要性。如前文对CITE-seq的研究所述,与RNA分子相比,蛋白质分子拷贝数的增加通常会导致对蛋白质特征的更强检测。因此,在CITE-seq中的蛋白数据可能代表了信息最丰富的模式,特别是在抗体panel全面代表了所有具有高特异性的细胞亚群的情况下。其他的panel可能遗漏了关键的或以前未发现的标记的抗体,或包含低结合特异性的抗体,在这种情况下,无监督性质的scRNA-seq可能是最有信息含量的。即使在相同的数据集中,每个模式定义细胞状态的相对效用也可能因细胞的不同而不同。
因此,我们设计了一个分析解决方案来实现这些目标,而不需要用户预先了解每种模式的重要性。我们首先在之前生成的8,617个脐带血单核细胞的CITE-seq数据集上介绍并演示我们的解决方案,该数据集包含10个免疫表型标记。对RNA和蛋白数据的独立无监督分析显示,细胞分类基本一致(图1A, B;补充图1),但确实显示了一些差异。例如,在分析转录组时,CD8+和CD4+ T细胞部分混合在一起,但在蛋白数据中清晰分离。相比之下,传统的树突状细胞(cDCs),以及罕见的红系祖细胞和小鼠类3T3对照,在分析RNA时形成不同的簇,但根据表面蛋白丰度与其他类型的细胞混合。有了生物学背景后,不同模式的细胞类型特异性差异可以通过CITE-seq panel的组成来预测,该panel包含抗cd4和抗cd8抗体,但缺乏任何免疫表型标记来区分cDCs。

使用加权最近邻分析的多模态积分概述
- (A, B)对来自CITE-seq脐血单核细胞数据集的转录组(A)和蛋白(B)模式的独立分析。蓝点在(A)和(B)中标记了相同的靶细胞。红点表示根据转录组(A)或蛋白(B)方式与靶细胞最接近的k=20个相邻细胞。
- (C)将相邻的RNA平均起来,以预测目标细胞的分子含量,并与实际测量值进行比较。由于RNA邻居代表了不同的T细胞亚群的混合物,CD4和CD8的预测和测量的蛋白表达水平存在很大的误差。
- (D)与(C)相同,但表示平均蛋白质邻居。由于邻近的蛋白质都是CD8 T细胞,所以预测值与实际测量值非常接近。因此,我们可以推断,对于这个目标细胞,蛋白质数据对定义细胞状态是最有用的,并赋予它一个更高的蛋白质模态重量。如补充方法中所述,我们在低维空间中进行预测和比较步骤。
- (E)我们可以通过构建一个基于蛋白质和RNA相似性加权平均值的加权最近邻(WNN)图来整合这些模式。此图的UMAP可视化和聚类。
- (F)数据集中所有细胞类型的RNA和蛋白模态权重的中位数。在不了解细胞类型标签的情况下计算每个细胞的模态权重。
对于每个细胞,我们首先对每个模态分别计算k=20个最近邻的集合。我们发现,对于CD8+ T细胞,最相似的RNA邻居往往反映了CD8+ T细胞和CD4+ T细胞的混合(在RNA KNN图中,总共有944条不正确的边连接CD8+和CD4+ T细胞)。相比之下,相邻的蛋白被正确地识别为CD8+ T细胞(在蛋白KNN图中,识别了12个CD8+/CD4+边)。这反映了蛋白质数据在定义这些细胞状态时的特殊效用。接下来,我们分别对蛋白质邻居和RNA邻居的分子谱进行平均(即预测相邻细胞的分子含量),并将平均值与原始测量值进行比较。我们发现,对于CD8+ T细胞,基于蛋白knn的预测比基于RNA knn的预测更准确。
然后,我们利用这些预测的相对准确性来计算RNA和蛋白质的模态“权重”,描述每个细胞的相对信息内容。我们在补充方法中为WNN工作流程的每个组成部分提供了详细的数学描述,并强调了三个关键步骤:
- 1)获得模态预测和跨模态预测,
- 2)基于细胞特定带宽核(cell-specific bandwidth kernel)将这些预测转化为预测亲和力,
- 3)使用softmax变换计算模态权重。RNA和蛋白质模态权重是非负的,对每个细胞都是唯一的,总和为1。
插播softmax解释:
我又翻出从未看过的花书《深度学习》,果然在第六章前度回馈网络(或多层感知机)中看到了相同的单词:softmax。
在机器学习尤其是深度学习中,softmax是个非常常用而且比较重要的函数,尤其在多分类的场景中使用广泛。softmax把一些输入映射为0-1之间的实数,并且归一化保证和为1,因此多分类的概率之和也刚好为1。
顾名思义,softmax由两个单词组成:soft max。对于max我们都很熟悉,比如有两个变量a,b。如果a>b,则max为a,反之为b。用伪码简单描述一下就是 if a > b return a; else b。
另外一个单词为soft。max存在的一个问题是什么呢?如果将max看成一个分类问题,就是非黑即白,最后的输出是一个确定的变量。更多的时候,我们希望输出的是取到某个分类的概率,或者说,我们希望分值大的那一项被经常取到,而分值较小的那一项也有一定的概率偶尔被取到,所以我们就应用到了soft的概念,即最后的输出是每个分类被取到的概率【引自网络】。
这个函数的作用就是提示我们在WNN的框架中把多模态和机器学习的多分类问题联系到一起。
我们的最后一步整合模式来创建一个“加权最近邻”(WNN)图。对于每个细胞,我们计算一组新的k-最接近的细胞,基于一个反映标准化RNA和蛋白质相似性的加权平均值的指标(补充材料方法)。神经网络图是多模态数据集的单一表示,但应该更准确地反映这两种数据类型的丰富性。例如,WNN图只包含20条CD8+/CD4+边。此外,许多常见的单细胞数据分析任务——包括tSNE/UMAP可视化、聚类和轨迹推断——都可以接受用户指定的邻接图作为输入。因此,我们使用我们的WNN图来推导一个集成的UMAP和我们的CITE-seq数据集的聚类(图1E)。与上述两种方式的单独分析不同,我们的联合整合清晰地分离了CD4+和CD8+ T细胞,保留了cDCs的身份,还发现了NK细胞内部细微异质性的其他来源(补充图1)。我们观察到,分类为CD8+ T细胞的细胞被赋予了更高的蛋白模态权重,而DCs被赋予了更高的RNA模态权重,尽管模态权重的计算是不受监督的,而且不知道细胞类型标签,但我们还是重申了我们的生物学预期类型(图1F)。
WNN分析是一种鲁棒且灵活的多模态分析方法
接下来,我们进一步探索了WNN集成的性能,评估了其对数据质量波动的稳健性,并对其他最近开发的方法进行了基准测试。为了进行这些分析,我们使用了最近生成的人类骨髓的CITE-seq数据集,代表30,672个单核细胞和25个抗体。虽然样本包含的细胞横跨整个范围的造血分化,抗体面板被设计用于分离组末期分化的细胞。
与我们之前的例子一致,WNN的整合大大提高了我们解决造血细胞状态的能力(图2A;一旦通过整合的多模态聚类对细胞状态进行注释,我们就能够发现各组中差异表达(DE)基因和蛋白,进一步验证其生物学特性和意义(补充图2)。然而,虽然这些细胞类型是由RNA和蛋白标记定义的,但单独进行非监督分析的统计力不足以识别这些群体,这也说明联合分析的重要性。的确,当检查细胞特异性模态权重时,我们发现t细胞组——特别是在scRNA-seq分析中被掩藏的群体——都获得了较高的蛋白模态权重(图2B)。

WNN的基准测试和鲁棒性分析。
(A)对CITE-seq数据集(人类骨髓单核细胞和25种表面蛋白)的分析。UMAP可视化使用RNA、蛋白质或WNN分析计算。细胞注释来自WNN分析,揭示了T细胞和祖细胞内的异质性,而这是两种模式都无法独立发现的。补充图2中显示了细粒度注释,它在分析一种模态时更清楚地表明性能欠佳。
(B)单细胞蛋白形态重量。祖细胞群体均接受低蛋白权重,而T细胞群体接受高蛋白模式权重,这与为分化细胞类型量身定制的抗体panel组成一致。
(C)为了检验WNN的鲁棒性,我们在蛋白质数据中加入了越来越多的高斯噪声。当噪音水平增加时,所有细胞的蛋白质重量都减少到0。
(D, E)将WNN与totalVI和MOFA+进行基准测试。
(D) WNN定义的整合潜在空间最准确地重构了25个蛋白的表达水平。
(E)与竞争方法相比,WNN分析减少运行时间。补充图2中提供了其他的基准测试分析。
相反,RNA权重最高的细胞群代表造血祖细胞群。这与我们的蛋白panel的组成是一致的,我们的蛋白panel没有包含区分多能和系承诺的CD34+祖细胞的表面标记。因此,我们的多模态分析能够识别不同群体的造血干细胞、淋巴启动多能祖细胞(LMPP)、红细胞祖细胞、血小板祖细胞、单核细胞祖细胞、B细胞祖细胞和常规/浆细胞样DC系,这些可在scRNA-seq数据中验证。我们确认了在10到50之间改变参数k(默认设置为20)只会对模态权重产生微小的差异(补充图2),这表明我们的结果对于该参数的变化是稳健的。

这些结果表明,WNN分析可以提供必要的灵活性,并允许一种数据类型弥补另一种数据类型的弱点。我们通过模拟实验证实了这一点,我们在ADT数据中加入了越来越多的随机高斯噪声,以模拟非特异性结合的增加(图2C)。我们发现,ADT噪音的增加导致了所有细胞类型蛋白质权重的下降,且呈剂量依赖性。此外,添加足够数量的蛋白质噪声后,蛋白质模态权重被分配为0,正确地指示下游分析只关注scRNA-seq数据。
接下来,我们将WNN与最近引入的两种多模态集成方法进行比较:多组因子分析 ( Multi-omics factor analysis,MOFA+),它使用基于因子分析的统计框架以及totalVI,它将深度神经网络与层次贝叶斯模型结合在一起。这两种方法都将模式整合到一个潜在空间中,我们利用这个潜在空间构建了一个集成的k-NN图和一个二维UMAP可视化。我们认为,我们可以通过比较每个细胞的分子状态与潜在空间中最邻近细胞的相似性来量化不同方法的性能。我们发现,对于25个蛋白中的每一个(图2D)以及RNA转录组(补充图2),WNN分析显示出优于或等效的性能。对于调节性T细胞(CD25)和效应性T细胞(CD57)的标记,表现上的差异尤其显著。这与UMAP可视化是一致的,在UMAP可视化中,WNN是唯一不将这些种群与其他种群混杂在一起的方法(补充图2)。WNN分析在速度上也有显著提高,在分析完整数据集时可达到15倍(图2E)。
虽然我们主要在CITE-seq数据集上演示我们的方法,但我们的策略适用于多种多模态技术。例如,最近的发展使得能够同时测量单核的ATAC-seq和转录组数据。我们将WNN分析应用于由10x Genomics Multiome ATAC+RNA kit生成的11,351对PBMC剖面数据集。我们发现,这些模式的组合显示出分离免疫亚群的最大能力(补充图3)。有趣的是,类似于CITE-seq分析,我们发现ATAC-seq数据更能分离初始CD8 +及CD4 + T细胞状态由于可靠的检测细胞特定类型开放的染色质区域(补充图3)。

ATAC和RNA数据的结合也使我们能够识别在我们的wnn定义的簇之间不同可达的DNA序列基序。例如,我们发现MAIT细胞中可达的ATAC-seq峰高度富集了促炎转录因子RORC的基序,在这些细胞中,RORC的转录也被上调(补充图3)。我们得出这样的结论:算法的分析能够敏感和强劲描述异质性,可灵活地应用于多种数据类型,进行综合多模态分析。
人类外周血单核细胞的多模态图谱
虽然流式细胞术和CyTOF被广泛用于对免疫细胞蛋白表达进行高维测量,但CITE-seq使用独特的寡核苷酸条形码序列提供了一个独特的机会,可以在细胞转录组的同时分析非常大的抗体panel。此外,我们最近证明,每个抗体的信噪比可以作为抗体浓度的函数优化任何单个标记,并表明CITE-seq数据质量不会随着总抗体的增加而下降。因此,我们策划并优化了一组包含228个抗体的TotalSeqA试剂(补充表1,略),其中包括一系列不同的谱系和激活标记。对于大型的CITE-seq ,一个潜在的担忧是,来自少数标记的信号可能会使整个文库饱和,大大增加了所需的测序深度。我们开发了一个简单的实验过程来解决这个(补充方法,略),首先确定12 228标记收到过多的读入初步实验,然后补充与非结合的panel(barcode-free)抗体小组为了抑制信号的大小。
我们利用CITE-seq技术以及优化的抗体panel和整合的WNN分析策略,生成人类PBMC的多模态图谱。我们从8名参与艾滋病毒疫苗试验的志愿者中获得了PBMC样本,年龄跨度20-49岁(中位年龄36.5岁)。每个受试者在三个时间点采集PBMCs:注射vsv载体HIV疫苗前(第0天)、第3天和第7天(图3A)。整个数据集由24个样本组成,并使用“Cell hash”来最小化技术批次效应。对于每个样本,我们使用10X Chromium 3 '(使用228 TotalSeq A抗体)对细胞进行分析,总共代表了161,764个细胞(平均8,003个独特RNA分子/细胞,5,251个独特ADT/细胞)。我们还使用ECCITE-seq对所有样本中共49,147个细胞进行了剖分分析,该技术可使用10X 5 '技术对表面蛋白进行。虽然后一组实验包含了54种抗体,其中包括实验室偶联抗体和TotalSeq-C试剂,反映了在实验时商业偶联的可用性,但我们也能够对这些细胞进行免疫库图谱分析。经过NovaSeq测序、严格的质量控制和双重过滤(补充方法),我们最终的数据集包含210,911个细胞,并允许我们分析静息(未接种)和激活(接种后)免疫系统的细胞异质性。

人类PBMC的多模态图谱
(A) CITE-seq实验设计原理图。PBMC样本来自8名志愿者接种前(第0天)和接种后(第3天和第7天)。我们使用10x3 '(228个抗体)和10x5 '(54个抗体+ BCR +TCR)技术对每个样本进行CITE-seq处理,共产生210,911个细胞。
(B-D)基于RNA数据(B)、蛋白数据(C)或WNN分析(D)对161,764个10x3 '细胞进行UMAP可视化分析。
利用WNN图的无监督聚类来识别细胞类型,并将其分成3个注释层,从8个大类到57个高分辨率聚类。49,147个10X 5 '细胞的UMAP可视化,映射到3 '参考数据上,如图6所示。
我们应用了“基于锚的”工作流,首先将样本整合在一起,使细胞能够基于它们共享的生物状态聚集在一起,而不是原始样本(补充方法,Seurat的FindAnchors)。虽然这导致未接种和已接种的样本最初聚集在一起,但它使我们能够在所有样本中一致地注释细胞状态,并在下游分析中了解细胞类型的特定反应。然后,我们使用WNN集成对两种模式进行联合分析,作为比较对照,分别使用RNA和蛋白模式对数据集进行可视化(图3B-D)。
我们在WNN分析中鉴定了57个簇,封装了所有主要和次要的免疫细胞类型,并揭示了显著的细胞多样性,特别是在淋巴系中。除了罕见的细胞类型外,每一簇细胞都来自全部24个样本。我们的聚类可以容易分为大类别,包括CD4 + T细胞(12集群),CD8 + T细胞(12群),非传统的T细胞(7群),NK细胞(6群),B细胞,浆细胞和plasmablasts群(8),树突细胞和单核细胞(8集群),和罕见的集群造血祖细胞、血小板、红细胞和循环先天淋巴细胞(ILC)。为了帮助解释聚类结果,我们为每个细胞分配了三个粒度越来越大的注释(级别1,8个类别;第2级,30个类别;3级;57个类别)。虽然在T细胞亚群有较大程度的异质性,我们的分析明确确定异构子集的髓细胞与最近的高分辨率scRNA-seq完全整合分析排序的数量,包括极其罕见的人群(0.02%)定义的树突状细胞表达 AXL 和SIGLEC6(ASDC;补充图4)。我们还发现炎症基因如IL1B和CCL3在单核细胞群体中的表达存在很大的异质性,但由于这种异质性在不同的志愿者中存在差异,因此我们保守地没有进一步细分这些状态(补充图4)。

接下来,我们对每个簇鉴定了差异表达的RNA和免疫表型标记。我们发现,对于两种模式,每个簇显示出不同的分子模式和生物标记(图4A中CD4+和CD8+ T细胞状态的热图。此外,这些确定的生物标记在人类志愿者和疫苗接种时间点之间是不变的。在多个样本中观察到一致的聚类生物标记提供了证据,表明我们观察到的异质性是生物学上可重复的,而不是由过度聚类驱动的。我们得出这样的结论:集成的无监督发现和注释是至关重要的,但一旦这些状态被测到,监督鉴别分析能够敏感地描述标记,从而定义他们的分子状态。
免疫细胞状态的多模式生物标志物
(A) CD4+ T细胞状态热图。标记包括通过差异表达(DE)鉴定的最佳RNA和蛋白特征。Heatmap显示了按细胞类型、供体和接种时间点对细胞进行分组的假体平均值,并表明标记物在不同PBMC样本之间不存在差异。
(B)与(A)相同,但对于CD8+ T细胞状态。附加图5显示了额外的热图。
(C)对于57个聚类中的每一个,我们都基于CITE-seq数据计算出了最佳的表面标记富集panel。柱状图显示了panel在模拟( in silico)中对每种细胞类型富集的能力。各panel的组成见补充表2。
(D) CD8_TEM_5集群预测标记面板的验证。我们根据(C)中识别的标记板对细胞进行分类,并进行大容量rna测序。根据我们的CITE-seq数据集,Heatmap根据预期要被DE的基因排序,并通过大容量RNA-seq进行验证。
(E)与(D)相同,但是为CD4 CTL细胞。
由于在CITE-seq中对蛋白特征的检测比较稳健,并且结合我们抗体panel的大小,我们认为我们可以发现每个簇的免疫表型标记的小panel,例如,可以通过流式细胞术进行靶向富集。我们使用逐步变量选择和logistic回归(补充方法)来为每个子集确定不同大小(1-10个标记)的最佳抗体标panel,并计算模拟( in silico)富集水平(图4C)。我们发现,单个标记能够对45个聚类实现至少10倍的有效富集,而有3个标记的panel足以对55个聚类实现10倍的富集。
这种标记发现程序可以识别特征良好的群体(浆细胞样DC (pDC): CD123+, MAIT细胞:CD3+ CD161+ TCRv (2) 7.2+, CD4 Naive: CD4+ CD45RA+ CD45RB+)的有效panel。在其他情况下,例如在CD4+淋巴细胞的细胞毒性种群中,我们的分析发现CD43是一个之前没有报道过的具有高富集能力的标记物。对于这一人群以及高细胞毒性CD8+ T细胞(CD8_TEM_5)亚群,我们通过常规流式细胞术和大容量RNA-seq(补充方法)在一组独立的健康供体PBMCs中成功验证了我们的富集panel。在这两种情况下,我们检查了根据我们的CITE-seq数据预计将被DE的基因表达水平,并且我们观察到在排序的剖面和CITE-seq簇之间有明确的一致(图4D, E)。
值得注意的是,我们的流式细胞术实验使用了在CITE-seq实验中所代表的精确抗体克隆,这有助于确保两种分析的结果一致。我们在补充表2(略)中报告了每一个panel,以促进对数据集中其他集群的类似实验。我们注意到,虽然这些panel可以实现高水平的富集,即使是最佳分类组可能包含少数来自其它污染细胞。我们在补充图4中展示了每个panel的精度和召回指标,表明使用少量标记对高分辨率子集的真正“同质”群进行分类仍然具有挑战性。
淋巴群内的多模态异质性
我们整合的WNN分析揭示了丰富的T细胞状态的多样性,这些T细胞状态通常在scRNA-seq分析中没有被捕获,包括CD4+调节性T细胞、MAIT细胞、休眠和双负性T细胞的多个亚群,以及初始、记忆和效应状态的异质性亚群。即使对于之前被流式细胞仪或CyTOF描述过的人群,我们的数据集也代表了一个强大的资源,用于描述每个细胞状态的丰富RNA和蛋白生物标记物。然而,我们也观察到以前在外周血中没有明确的异质性来源。
例如,在CD8+记忆T细胞中,我们通过整合素蛋白CD49a和CD103的双峰互排式(bimodal and mutually exclusive)表达确定了不同的亚群(图5A)。虽然我们在外周血中发现了这些细胞,但这些蛋白的表达传统上与组织常驻记忆(TRM)细胞密切相关,其中整合素有助于介导对上皮细胞或细胞外基质的黏附。CD8+ CD103+ T细胞表面表达高水平的异二聚体共结合伙伴整合素-7(图5B),而在CD8+ CD49a+组中无表达。我们通过对相同标记物进行流式细胞术,验证了独立健康PBMC样本中人群的存在(图5C, D)。此外,我们鉴定了这两组间差异表达基因的模块(图5E),这些模块被富集用于T细胞激活、分化、信号应答和趋化模块(图5F)。两个种群都没有表达典型的常驻标记CD69(补充图6),这表明它们不是暂时脱离并重新进入循环的TRMs。相反,这些亚群可能代表准备成为组织驻留的细胞,并且已经开始获得区别的分子特征。

描述淋巴群内的异质性
(A) CITE-seq检测CD8+ T记忆细胞内整合素蛋白CD103和CD49a的互斥表达。
(B) CITE-seq检测CD103+ CD49a-和CD103+ CD49+群体整合素-7的差异表达。
(C-D)流式细胞仪验证了这些人群的存在。图与(A-B)相同,但由流式细胞仪生成。
(E-F) CD103+ CD49a-和CD103- CD49+群体间差异表达基因,并GO富集。
(G)点图显示数据集中15个最丰富的T细胞克隆类型的表示。对于空间,只有VDJ区域显示在y轴上,但是克隆中的所有细胞共享相同的CDR3序列。克隆存在于有限的细胞毒性和效应细胞状态,并在接种时间点之间共享。每个点的大小代表克隆型细胞的数量。被归类为cmv阳性的捐赠者体内的克隆体呈红色。
(H)克隆细胞表现出类似的分子特征。灰色点代表T细胞,使用10X 5 '法测定TCR序列。
(I-K) NK细胞的异质性是由与CD16和CD38蛋白表达相关的两个梯度决定的。
(I) NK细胞是根据CD16蛋白的定量表达来排序的。与CD16正相关或负相关的基因表达的滚动平均值显示为平滑的线条。(J)与(I)相同,但是为CD38。
(K) CD38和CD16蛋白表达有两个独立的梯度,在NK细胞中不相关。
除了描述mRNA和蛋白表达的异质性,我们利用我们的5 '数据集来探索分子状态和TCR序列之间的关系。我们获得了代表16,060个不同克隆的高产TCR的细胞群/细胞群序列,其中一个克隆中的所有细胞共享完全相同的CDR3和CDR3序列的细胞群。整个克隆多样性在接种时间点上是一致的,与预期7天内接种疫苗后没有淋巴反应一致,97%的克隆只包含一个单细胞。然而,我们也观察到扩增的克隆群体的存在。作为阳性对照,我们观察了高度限制使用TCRa序列的人群:在多个志愿者中,MAIT和不变NKT细胞都表现出了密切相关的转录谱和半不变库(补充图6)。

除这些群体外,我们还鉴定了至少包含10个细胞的31个扩增克隆(图5G)。在每个病例中,克隆群体中的细胞都表现出极其相似的分子特征(图5H),代表CD8+ T细胞亚群(主要在我们之前鉴定的CD8_TEM_4和cd8_tem_5簇内)以及细胞毒性CD4+ T细胞(CD4 CTL)。每个克隆通常代表一个志愿者的细胞,但是可以在多个时间点独立发现,包括接种疫苗之前(图5G)。由于我们的样本志愿者一般都是中年人,身体健康,我们认为过度繁殖的克隆可能与巨细胞病毒(CMV)感染有关。我们通过CMV肽池刺激PBMC并进行细胞内细胞因子染色来评估每个志愿者的CMV状态,以确定CD8+ T细胞的反应(补充方法,补充表3,略),确定5个阳性和3个阴性志愿者。我们发现5个阳性样本占扩增克隆细胞的91%。
我们注意到,虽然WNN整合提高了发现不同细胞亚群的能力,同时它也可以改善细胞轨迹和连续异质性来源的特征。例如,在B细胞中,我们确定了一条由规范蛋白标记IgD和CD27定义的连接幼稚细胞和记忆细胞的连续轨迹以及一个相关基因模块(补充图6)。类似地,NK细胞被细分为五个簇,代表了连续景观的变异。我们的数据显示,传统的将NK细胞分为CD56-bright和CD56-dim两类,这代表了CD16表达定义的更广泛的连续统一体以及调节细胞毒性并与该标记物正负相关的基因模块(图5I)。
我们还观察到CD38表达定义的第二个梯度,据我们所知,这是以前没有描述过的。值得注意的是,编码NK活化受体NKG2C的KLRC2与这个连续体呈负相关,而信号转导适配器FCER1G则与这个连续体呈正相关(图5J)。这种表达模式与在CMV血清阳性个体中观察到的“适应性”或“记忆样”NK细胞的发展是一致的。值得注意的是,当我们的分析仅限于CMV T细胞应答阳性或阴性的个体时,我们观察到了一致的趋势(补充图6)。我们还在一个独立的人类PBMC[48]的CITE-seq数据集中观察到一致的结果。我们的结果表明,这种表型并不代表一个严格的非此即彼的现象,可能不是特异性的巨细胞病毒反应。最后,我们观察到CD38和CD16表达之间的最小相关性(图5K),表明NK细胞沿这些标记物定义的二维梯度下降。
综上所述,这些结果表明我们的数据集代表了一个强大的资源,可以枚举免疫系统中的细胞状态,确定细胞类型特异性富集的最佳试剂,并了解在克隆相关或抗原特异性细胞群中的分子异质性。由于这些结果在接种前和接种后的时间点上是一致的,它们可能描述了健康免疫系统的一般特征。我们已经发布了一个门户网站https://atlas.fredhutch.org/nygc/multimodal-pbmc,以促进社区对该资源的探索。
对疫苗的最初先天反应的特征
接下来,我们探索了我们的数据集,以描述我们之前确定的每个细胞类型对疫苗接种的反应。我们特别感兴趣识别细胞的数量造成影响最强烈的先天免疫反应,预计将高度激活第一次接种疫苗的时间点(第三天),随后抑制在第二个时间点(第七天)与另一个non-replicating HIV病毒载体疫苗。我们对表现为细胞类型特异性改变的反应感兴趣,无论是在基因表达,蛋白表达,或丰富的免疫接种时间点。
为了量化每个簇的反应强度,我们首先确定了每种细胞类型在接种前和第3天细胞间差异表达的基因和蛋白的数量。然而,由于pergene测试对每个集群中的细胞数量高度敏感,我们还利用了另一种方法(“扰动评分”,perturbation score),该方法保留了在完整转录组的基础上从不同时间点区分细胞的统计能力。正如预期的那样,我们观察到强劲反应骨髓亚种群的一个子集,但只有最小反应淋巴组织(图6 a、B)。基本上也是一致响应模式样本,除了一个志愿者展示一个高度激活免疫系统在接种之前,这是进一步分析(补充图7)。
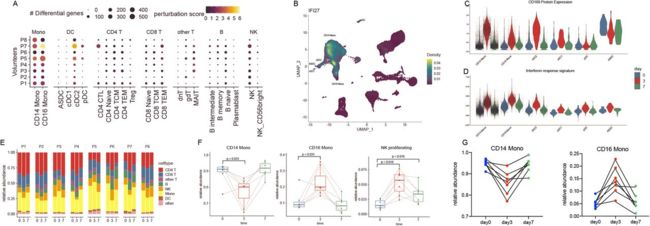
识别对疫苗接种的细胞型特异性反应
(A)对于每个2级标注的细胞簇,我们计算未接种(第0天)和第3天样本之间差异表达基因的数量(每个点的大小)。由于每个基因测试对细胞数量高度敏感,我们还计算了一个“扰动分数”,它反映了基于整个转录组(每个点的颜色)的分子反应的强度
(B)由星云包制作的密度图,显示典型干扰素应答基因IFI27的表达
(C) Violin图显示第3天样品中单细胞中siglecl -1 (CD169)蛋白的上调,以及干扰素应答的签名
(D),在选择的细胞类型。在(A-D)中,我们始终观察到仅在CD14+单核细胞、CD16+单核细胞和cDC2 DC中观察到强劲的反应。
(E) Barplot显示,在整个疫苗接种过程中,大组(1级注释)的频率是稳定的。
(F)在这些大类中,免疫接种期间经典单核细胞、非经典单核细胞和增殖NK细胞的相对丰度。p值是使用配对Wilcoxon检验计算的。
(G)流式细胞仪测定的单核细胞群体的相对丰度。
我们在CD14+经典和CD16+非经典单核细胞中观察到最强的变化,因为两种细胞类型上调了62个高富集基因的共享模块,这些基因对I型干扰素反应良好(图6A, B;补充图7)。此外,我们鉴定了siglec1 (CD169)作为蛋白反应的生物标志物,它只在第3天的样品中被显著诱导(图6C)。当我们检测树突状细胞群时,我们观察到只有在CD1C+ cDC2细胞中才有类似的强劲反应。相比之下,CD141+ cDC1、ASDC和pDC的密切相关人群表现出了最小的反应,我们在接种前和接种后都没有检测到任何DE基因(图6A)。这说明在DC亚群中,cDC2s可能在该疫苗应答过程中下游的适应性免疫系统的启动和激活中发挥重要作用。

接下来,我们试图确定细胞类型在处理时间过程中表现出的变化。我们没有观察到广泛免疫类总体丰度的显著变化(图6E;级别1的注释),因此我们关注于识别这些更广泛的组中更细微的组合变化。例如,虽然单核细胞的总体比例在各个时间点上是一致的,但是在第0天和第3天之间,经典细胞和非经典细胞的比例出现了强烈的变化(图6F)。我们验证了这一结果,在第7天观察到的回归基线比率,使用流式细胞术对相同的样本(图6G)。我们没有观察到淋巴样细胞内的变化,只有一个例外:一小群NK细胞表达增殖和细胞周期基因(NK_proliferation),在接种疫苗后持续增加(图6F)。这些发现在3’和5’scRNA-seq实验的独立分析中是可重复的,并且在第3天和第7天的样本中都持续存在(图6F这一发现可能反映了NK细胞发育和成熟的早期步骤,NK细胞是NK细胞介导的先天免疫反应[52]的关键成分。
将查询数据集映射到多模态参考数据集
免疫系统的单细胞转录组分析已成为常规,不仅适用于健康受试者,也适用于多种临床情况,包括COVID-19患者。这些数据集通常使用一个工作流处理,该工作流由无监督集群和随后的细胞类型注释组成。这个过程假设最小的先验知识,是理想的适合发现细胞类型。在构建了免疫系统的多模态参考后,我们试图利用该数据集来协助分析和解释其他分析人类PBMC(查询)的单细胞实验,即使只有转录组数据。
我们首先对参考数据集中的转录组测量应用一种称为“监督主成分分析(sPCA)”的程序。sPCA不像PCA那样寻找一个能最大化总方差的低维投影,而是识别一个转录组数据集的投影,最大限度地捕捉WNN图中定义的结构。形式上,给定一个基因表达矩阵X和一个WNN图Y, sPCA确定了在UTX和Y的线性核之间最大化Hilbert-Schmidt独立性标准测度的变换矩阵U(补充方法,略)。sPCA允许加权转录组和蛋白质测量来帮助“监督”基因表达数据的分析,并确定在我们的多模态参考中定义细胞状态的最佳转录组载体(基因模块)。
我们在我们的参考(其中mRNA和蛋白质同时被测量)上计算这个sPCA转换,但是随后可以快速地将这个转换投影到任何scRNA-seq查询数据集上。将这种转换与前面描述的基于“锚”的框架结合起来,允许我们将每个scRNA-seq查询细胞放在前面定义的引用UMAP可视化(补充方法,略)上,并基于引用集群对其标识进行注释。我们注意到与我们之前开发的scRNA-seq集成算法相比,在此过程中,参考数据集和可视化可以保持不变。
与无监督分析相比,这种有监督的映射过程极大地提高了我们分析和解释查询scRNA-seq数据集的能力。我们在流感疫苗接种前检查了最近生成的人类PBMC数据集,该数据集测量了53,099个细胞和82个表面蛋白的转录组。我们这个数据集映射到参考只使用转录组数据和转移我们的2级注释,揭示存在的多个高分辨率淋巴(补充图8)子集。我们验证的准确性预测使用查询蛋白质数据,举行的引用映射过程,然而表达模式基于我们预测注释透露,充分整合与参考数据集。例如,注释为调节性T细胞的细胞在CITE-seq数据中表达CD25+,我们观察到MAIT细胞(CD161+)、memory (CD45RA- CD45RO+)和naive (CD45RA+ CD45RO-) T细胞以及循环ILC (CD117+ CD25+,补充图8)也有类似的结果。我们以scArches(一种最近开发的将scRNA-seq查询映射到参考数据集的方法)为基准对我们的方法进行了测试,并观察到我们的方法在准确性和性能方面取得了显著的改进(图7A, B;补充图8)。

免疫扰动的监督映射
(A)小提琴图显示了我们的CITE-seq数据集中九个蛋白质的表达模式。根据wnn定义的T cell Level 2注释
(B)对人类PBMC的独立CITE-seq数据集中的相同蛋白质进行分组(Kotliarov et al., 2020)。细胞根据他们预测的基于转录体的参考映射的注释进行分组。蛋白质数据未被绘制,但显示出与(A)相同的模式。
(C) UMAP可视化(Wilk et al, 2020) scRNA-seq数据集,其中包括44,721名COVID-19住院患者的PBMC和健康对照。UMAP是使用无监督分析计算的。
(D)与(C)中相同,但在数据集已映射到我们的多模态参考之后。细胞由其预测的2级注释着色。
(E)基于scRNA-seq参考图(y轴)和CyTOF (x轴)对样本的MAIT细胞丰度进行量化(Wilk等,2020)。这两种方法之间的Pearson相关性为0.911。
(F) CyTOF定量COVID-19患者和健康对照PBMC样本中MAIT细胞丰度。p值是使用未配对的Wilcoxon检验计算的。
接下来,我们将绘图方法应用于最近的一项scRNA-seq研究,该研究分析了7名住院的COVID-19患者的PBMC样本,以及6名健康对照。最初的文章对完整的数据集进行了无监督的聚类,并识别出6个T细胞簇(3个CD4+ T, 2个CD8+ T,和2个CD8+ T细胞)。在我们的监督分析我们转移2级注释,成功将T细胞分成12组(图7 c, D)。值得注意的是,有区别的中性粒细胞,所确定的原文是唯一出现在COVID-19但在参考样品中不能很好地映射。
利用我们的监督注释来测试不同疾病条件下细胞类型丰度的差异。例如,我们的发现重复了最初的无监督分析,强调了在COVID-19反应期间plasmablast频率的增加(补充图8)。然而,我们也观察到细胞状态的比例变化,这些变化在无监督聚类中未被检测到,在参考映射后被成功注释。特别是,我们观察到与健康对照相比,COVID-19样本中MAIT细胞减少。为了验证我们的发现,我们对原始样本和16个额外样本的验证队列进行了CyTOF。在原始队列中,我们观察到scRNA-seq预测的和CyTOF测量的MAIT细胞比例有很强的定量一致性(R=0.911)(图7E)。此外,对更大样本集的CyTOF分析发现,COVID-19样本中MAIT细胞减少(图7F;这种丰度的变化可能反映了这些细胞在抗病毒免疫反应中退出循环,在屏障组织中发挥保护作用。

讨论
为了利用多种数据类型来定义细胞身份,我们开发了加权最近邻分析,可以学习每一模态的信息内容,并生成多模态数据的综合表示。通过计算细胞特有的模态权重,WNN分析解决了多模态数据集分析的一个重要技术挑战,并允许跨一系列模态和数据类型的灵活应用。我们在整个文章中证明,对数据类型的加权组合进行下游分析,极大地提高了我们描述细胞多样性的能力。
我们应用我们的方法来分析人类PBMC的数据集,该数据集具有成对的转录组和228种表面蛋白的测量,代表了免疫系统的多模态图谱。我们利用这一资源来描述以前仅用scRNA-seq观察不到的广泛的淋巴样异质性,包括整合素蛋白在循环记忆T细胞上的异质表达,NK细胞中适应性反应的梯度,以及效应组和细胞毒性组中紧密聚集的克隆群体。我们的数据还使我们能够探索先天免疫系统对疫苗接种的反应,突出特异反应的生物标志物以及传统DCs的异质反应。重要的是,我们证明了可以很容易地挖掘CITE-seq数据来为任何感兴趣的亚群识别最佳的免疫表型标记panel。这些标记板可以与我们的CITE-seq面板中相同的抗体克隆用于流式细胞术,促进这些组的快速富集和下游分析以挖掘我们数据的价值。
除了构造多模态参考数据集之外,我们还演示了将scRNA-seq数据映射到此数据集的能力。我们通过主成分分析的监督版本来确定最好的转录组模块来完成这一任务,这些模块描述了我们的wnn定义的细胞类型。监督映射代表了非监督分析的一个有吸引力的替代方案,我们展示了这种工作流程如何可以改进细胞类型鉴定和强有力地整合来自多个捐赠者和疾病状态的样本。为了帮助同行社区利用我们的资源,我们创建了一个web应用程序,可以在http://www.satijalab.org/azimuth免费获得,它可以让用户快速在线绘制他们自己的数据集,自动化可视化和注释过程。使用这种方法,50,000个单元的数据集可以在不到5分钟的时间内被完全处理和映射。随着各种疾病状态下人类PBMC的谱分析变得越来越常规,对这些数据集进行自动图谱的能力将促进复杂免疫反应的表征和致病人群的发现。
最后,我们注意到,在我们的过程中学习的模态权重不仅可以作为测量类型的技术质量的表征,而且还可以反映在决定细胞身份时每种模态的生物学重要性。例如,我们对人类骨髓的分析表明,祖细胞和分化细胞表现出不同的模态权重。未来技术趋向同时在单个细胞内测量中心法则包括染色质状态、DNA甲基化、转录、空间位置和蛋白质水平。WNN算法的分析有助于揭示细胞的亚种群差异如何利用这些方法来调节他们的当前状态和未来的潜力。虽然我们目前对WNN分析的实现侧重于对两种模式的分析,但随着这些技术的成熟,该框架可以很容易地扩展到处理任意数量的多模态数据。因此,综合多模态分析提供了一种途径,可以超越细胞的局部和转录聚焦的观点,并对细胞行为、身份和功能进行统一定义。
文章资源
Seurat v4是在开放源码GPLv3许可下发布的,所有代码都可以在www.github.com/satijalab/seurat上获得。安装说明、教程和文档可以在www.satijalab.org/seurat上找到
为本手稿生成的CITE-seq数据可在https://atlas.fredhutch.org/nygc/multimodal-pbmc/下载和探索。
为了便于将新的查询数据集映射到手稿中描述的多模态PBMC参考,我们发布了一个自动web应用程序,Azimuth: www.satijalab.org/azimuth
https://www.biorxiv.org/content/10.1101/2020.10.12.335331v1.full
https://zhuanlan.zhihu.com/p/105722023
https://blog.csdn.net/bitcarmanlee/article/details/82320853