参考这些
主要根据这篇操作!!!
我觉得学RNA-seq必看的文献!!
这篇写的特别好:RNAseq-workflow
RNA-seq原始数据的下载方法
fastq格式大全
本科生搞定RNA-seq上游数据分析
RNA-seq一般流程
转录组入门:序列比对
RNA分析简洁版
序列比对
我觉得这个作者这样的分类特别清晰,值得学
new_workflow/
│ └── annotation/ <- Genome annotation file (.GTF/.GFF)
│
│ └── genome/ <- Host genome file (.FASTA)
│
│ └── input/ <- Location of input RNAseq data
│
│ └── output/ <- Data generated during processing steps
│ ├── 1_initial_qc/ <- Main alignment files for each sample
│ ├── 2_trimmed_output/ <- Log from running STAR alignment step
│ ├── 3_rRNA/ <- STAR alignment counts output (for comparison with featureCounts)
│ ├── aligned/ <- Sequences that aligned to rRNA databases (rRNA contaminated)
│ ├── filtered/ <- Sequences with rRNA sequences removed (rRNA-free)
│ ├── logs/ <- logs from running SortMeRNA
│ ├── 4_aligned_sequences/ <- Main alignment files for each sample
│ ├── aligned_bam/ <- Alignment files generated from STAR (.BAM)
│ ├── aligned_logs/ <- Log from running STAR alignment step
│ ├── 5_final_counts/ <- Summarized gene counts across all samples
│ ├── 6_multiQC/ <- Overall report of logs for each step
│
│ └── sortmerna_db/ <- Folder to store the rRNA databases for SortMeRNA
│ ├── index/ <- indexed versions of the rRNA sequences for faster alignment
│ ├── rRNA_databases/ <- rRNA sequences from bacteria, archea and eukaryotes
│
│ └── star_index/ <- Folder to store the indexed genome files from STAR
前半部分是我的血泪史!正文在中间开始!
这里都是坑,别学!!!!正确的在下一条分界线那里↓
1.1 在SRA数据库下载数据
1.2 转为fastq格式
fastq-dump *.sra
到这里才完全发现问题!!!!!
我的数据其实是有问题的!!!
我以为是单端测序(SE),但是在NCBI上查询了一下原来是双端测序(PE)!!!

所以就是重头来过!!!!
▶所以这里才是正文的开始!!
1 原始数据下载
1.1 在SRA数据库下载数据
SRR_Acc_List.txt里面是要下载的SRR号
注意!!!!光标一定不能在有SRR号的那行,不然会跳过那行!!
cat SRR_Acc_List.txt | while read id; do (prefetch ${id} );done
1.2 sra转成fastq (这里要注意不是这么写!!!!
因为是双端测序数据!!!
fastq-dump *.sra 绝对不能这么写!!!!
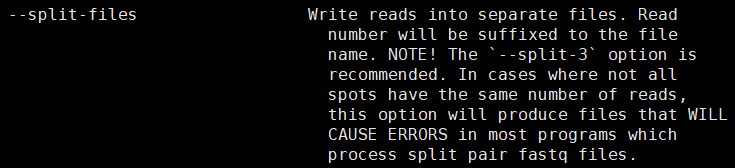
应该加这个参数 --split-files
fastq-dump --split-files *.sra
这里我会把.sra后缀的文件都移到一个新文件夹sra里,然后cd到sra文件夹里转fastq↓
cp ./SRR*/.sra ./sra
fastq-dump --split-files *.sra
如果需要压缩,可以加--gzip,就会生成.fastq.gz文件,解压缩用gunzip
单端测序SE用-U参数
而且不知道是不是我的错觉,好像分开两个文件的速度比合并在一个文件中速度快一些
1.3 md5sum检测传输的fastq文件是否完整,一般会附带一个MD5校验文件
md5sum *.fastq | tee md5sum.txt

2 质控
2.1 fastqc生成质控报告 fastqc.html和 fastqc.zip格式
fastqc -o . *.fastq
-o表示输出文件的位置

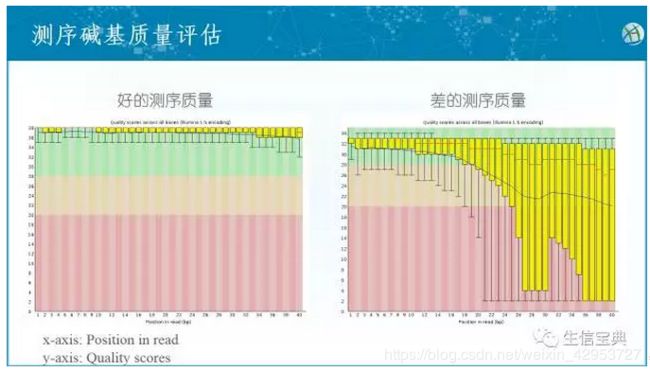
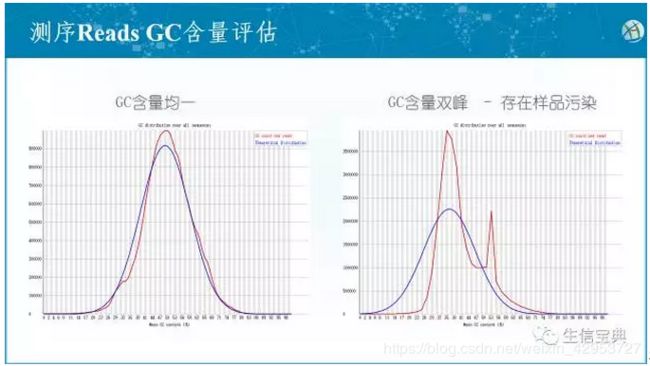
参考这篇
fastqc质控
multiqc将各个样本的质控报告整合为一个
multiqc *.zip
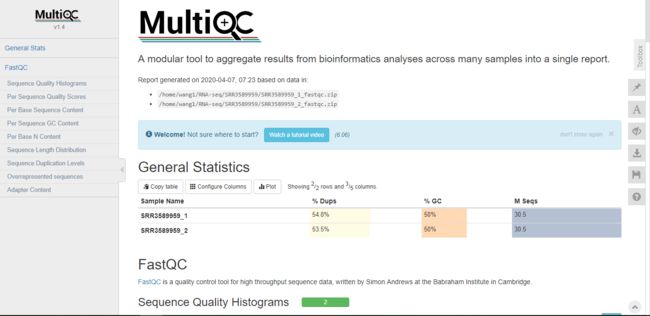
2.2 trim_galore修剪数据,用于去除低质量和接头数据
参数--fastqc:在数据过滤后再次质检
#需要新建一个clean文件夹
mkdir ./clean
cd ./clean
ls 你放fastq数据的路径/sra/*_1.fastq >1
ls 你放fastq数据的路径/sra/*_2.fastq >2
paste 1 2 > config
如果是单端就直接↓
ls fastq数据位置/*.fastq > config
#config数据里其实就是fastq文件的路径
#linux里输
cat config |while read id
do
#二选一
#双端
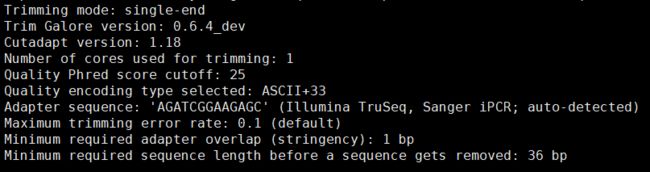
trim_galore ${id} -q 25 --phred33 --length 36 --stringency 3 --paired -o ./
#单端
trim_galore ${id} -q 25 --phred33 --length 36 --stringency 3 -o ./
done
#或者也可以写成.sh,然后
bash qc.sh config

3 比对到参考基因组
我们有时纠结是否真的需要比对:
如果你只需要知道已知基因的表达情况,那么可以选择alignment-free工具(例如salmon, sailfish),如果你需要找到noval isoforms,那么就需要alignment-based工具(如HISAT2, STAR)
打分机制:
reads比对到基因组上的一个位置,我们称之为一个alignment。 软件会对所有的alignments进行打分和判断,能够符合过滤条件的alignment称之为valid alignment, 只有valid alignments, 才会输出。
和BLAST类似,但在面对巨量的短序列数据时,类似BLAST这样的软件实在太慢了!因此,需要更加有效的数据结构和相应的算法来完成这个搜索定位的任务
每个alignment也有对应的打分机制。hisat2从以下几个方面对alignment进行打分
1 错配碱基罚分
错配碱基的罚分通过--mp参数指定,其值为逗号分隔的两个数字,第一个数字为最大的罚分,第二个数字为最小的罚分
2 reads上的gap罚分
gap的罚分通过分成两个部分,第一次出现gap的罚分和gap延伸的罚分,reads上的gap罚分通过--rdg参数指定,其值为逗号分隔的两个数字,第一个数字为gap第一个位置的罚分,第二个数字为gap延伸的罚分。
3 reference上的gap罚分
reference上的gap罚分通过--rdg参数指定,其值为逗号分隔的两个数字,第一个数字为gap第一个位置的罚分,第二个数字为gap延伸的罚分。
经过一系列的罚分机制,每个alignment会有一个对应的得分,然后会根据一个阈值,来判断这个得分是否满足valid alignment的要求。
hisat通过
--score--min参数指定该阈值,指定方式是一个和reads程度相关的函数,默认值为L,0,-0.2, 对应函数为f(x) = 0 - 0.2 * x
根据reads长度,可以计算出得分的阈值,大于该阈值的alignment 被认为是valid alignment , 才可能被输出。L代表线性函数,此外,也支持其他类型的函数,比如常量,自然对数等,更多选择请参考官方文档。
一条reads可能会拥有多个valid alignments, 在输出时,并不会输出所有的alignments, 而是只输出-k参数指定的N个alignments,-k参数的默认值为5。
输出结果以SAM格式保存,默认输出到屏幕上,可以通过-S参数指定输出文件。
hisat2 -t -x genome路径/genome -1 SRR路径/SRR3589959_1.fastq.gz -2 SRR路径/SRR3589959_2.fastq.gz -S 输出路径/SRR3589959.sam
-S表示输出sam文件
只需要写genome就好,它识别的是前缀,不是某一个文件!
hisat2 -t -x
但是报错了,需要re-run on a computer with more memory
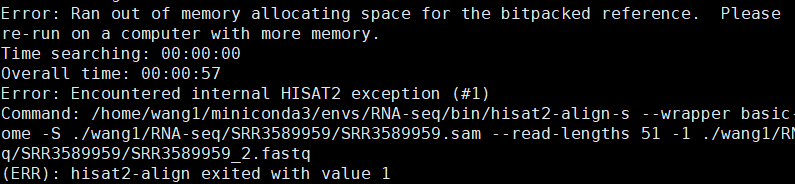
有人建议重新下载
index,于是我在Hisat2官网找到了index
hg19(grch37) 和 hg38(grch38) 的区别
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/grch38.tar.gz
tar -zxvf grch38.tar.gz
压缩包有make_grch38_tran.sh文件,详细记录了创建索引的过程。
但是还是报错,可能真的是内存不足
未完待续……
再更新:
也有可能是index的问题?
可是我这个index是直接从hisat2官网下载的啊……
我决定先放弃Hisat2,试试bowtie2
建立矩阵
bowtie2-build 基因组路径/hg19.fa index
但是建立不起来
试试STAR
参考这篇
STAR --runThreadN 6 --runMode genomeGenerate --genomeDir 你的路径/hg19/ --genomeFastaFiles 你的路径/.fa --sjdbGTFfile 你的路径/.gtf --sjdbOverhang 100
--runThreadN :设置线程数
--genomeDir:index输出的路径
--genomeFastaFiles :参考基因组序列
--sjdbGTFfile :参考基因组注释文件
--sjdbOverhang :这个是reads长度的最大值减1,默认是100
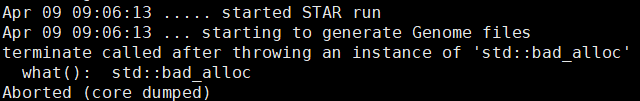
这位朋友说加一个加内存的参数
--limitGenomeGenerateRAM 80000000000(运行内存加到80G)
--genomeChrBinNbits 11 这个的计算方式 : log2((基因组长度)120,000,000,000/(NumberOfReferences)10,253,694)=11
但是还是不行,又看到大家的讨论,里面有这么一位网友↓

所以我觉得在我的小小虚拟机里应该是跑不动了,放弃 :-(
这篇作者在评论里建议用salmon
具体操作参考这篇
Salmon是Alignment-free transcript quantification;相比Alignment-based transcript quantification而言,省略了比对这一步骤(有些软件是只进行’轻度’比对),从而直接将reads分配到转录本上;后者相比前者spliced alignment能极大的减少计算机资源的使用
现在Salmon支持两种模式将readsmapping到转录本上:
第一种是quasi-mapping-based mode,其是一种最新的以及快速准确的定量转录本的方法,不需要像传统的那样需要通过比对才能将readsmapping到转录本上;
第二种则是alignment-based mode,跟类似RSEM软件一样,提供比对后的bam/sam文件即可对转录本进行定量
第一种方法需要对转录本建index,第二种方法则不需要
所以也许可以吧!
Salmon到这里下载
下好后放到src文件夹下
tar zxvf salmon-1.1.0_linux_x86_64.tar.gz
cd salmon-latest_linux_x86_64/bin
先试试第一种方法quasi-mapping-based mode:
- 建立索引 2. 对
reads进行生物学定量
salmon index -t 你的路径/hg19.fa -i 你的路径
index 代表建立索引
-t .fa文件的路径
-i 索引存放路径
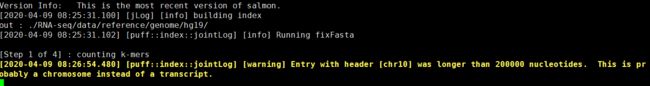
然后我突然明白,数据需要用转录本!!!!
……
后来又尝试了Prokka注释
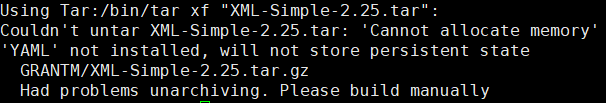
但是失败在了这一步。。。我真的放弃了:-(
2020.6.6更新
在实验室服务器上试试
#新建一个index文件存放index
cd ./index/
wget -O hg38.tar.gz https://cloud.biohpc.swmed.edu/index.php/s/hg38/download
tar -zxvf *.tar.gz
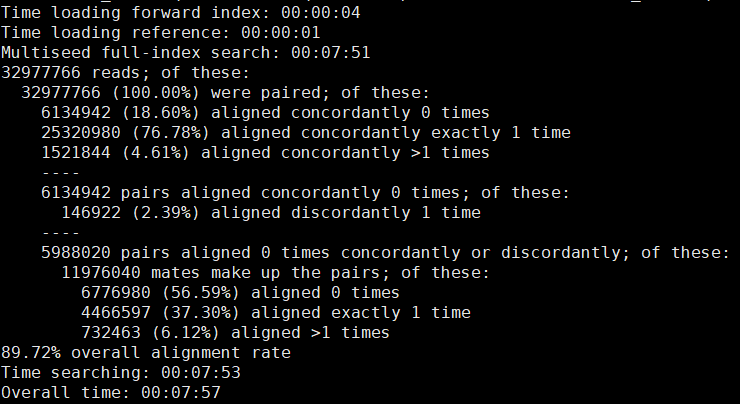
一条reads可能会拥有多个valid alignments, 在输出时,并不会输出所有的alignments, 而是只输出-k参数指定的N个alignments,-k参数的默认值为5。
输出结果以SAM格式保存,默认输出到屏幕上,可以通过-S参数指定输出文件。
hisat2 -t -x 存放index路径/index/hg38/genome路径/genome -1 SRR路径/SRR3589959_1.fastq -2 SRR路径/SRR3589959_2.fastq -S 输出路径/SRR3589959.sam
-S表示输出sam文件
只需要写genome就好,它识别的是前缀,不是某一个文件!
可以批量转,像我之前把.fastq都放在一个文件夹里了,并且是双端测序,所以↓
for i in `seq ? ?`
do
#将fastq文件转换成sam文件
hisat2 -t -p 8 -x index的路径/ctrl/index/hg38/genome -1 sra数据的路径/sra/SRR1234${i}_1.fastq -2 sra数据的路径/sra/SRR1234${i}_2.fastq -S SRR1234${i}.sam
done
这段可以直接在linux命令行运行,或者可以写成脚本运行
??表示两个数字,一个是起始数字,一个是终止数字,自己替换
4. Samtools: 转换为.bam格式+排序+索引
4.1 Samtools将.sam文件转为.bam文件
for i in `seq ? ?`
do
#将sam文件转换成bam文件
samtools view -S SRR59618${i}.sam -b > SRR59618${i}.bam
#对bam文件进行排序
#刚开始加了参数-o,运行后电脑终端一直不停跳乱码的东西,Xshell直接死机
samtools sort SRR59618${i}.bam > SRR59618${i}_sorted.bam
#对bam文件建立索引
samtools index SRR59618${i}_sorted.bam
done
4.2 对.bam文件进行排序sort
samtools sort SRR3589959.bam > SRR3589959_sorted.bam
4.3 对sorted.bam文件建立索引
samtools index SRR3589959_sorted.bam
运行后得到这三种文件
SRR3589959.bam
SRR3589959_sorted.bam
SRR3589959_sorted.bam.bai
#下载hg19_RefSeq.bed文件
$ cd /mnt/f/rna_seq/data/reference/genome/hg19
$ wget https://sourceforge.net/projects/rseqc/files/BED/Human_Homo_sapiens/hg19_RefSeq.bed.gz/download
#查看基因组覆盖率
$ read_distribution.py -i /mnt/f/rna_seq/aligned/SRR3589956.sorted.bam -r /mnt/f/rna_seq/data/reference/genome/hg19/hg19_RefSeq.bed
5. IGV可视化结果
参考这篇
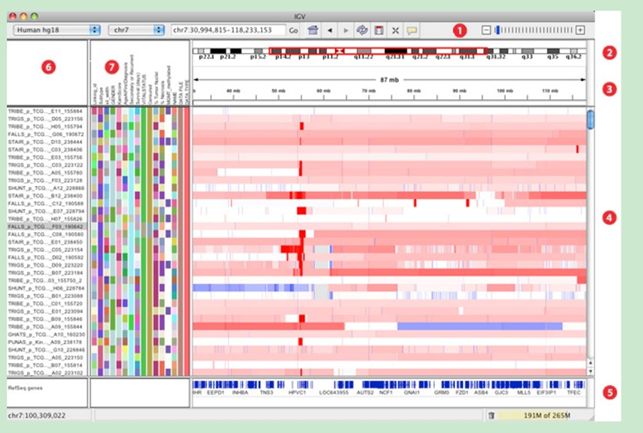
主窗口布局:
1.工具栏tool bar
2.红色框显示当前显示的染色体的位置,当缩小显示范围到整个染色体范围时,红色框消失。
3.显示当前查看的染色体序列的长度
4.该窗口显示测序样品的测序情况。每一条track代表一个样品或者一次实验,显示的情况包括甲基化、表达水平、拷贝数,碱基突变等信息。
5.参考基因组信息
6.track名(即样品或者实验名)
7.Attribute names属性名,即序列信息,如indel、甲基化等。
5.1 选择Genomes→load genomes from file 导入下载好的 .fa基因组

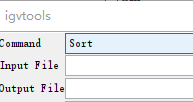
5.2 载入基因组注释,选择Tools→Run igvtools,进入以下igvtools窗口:

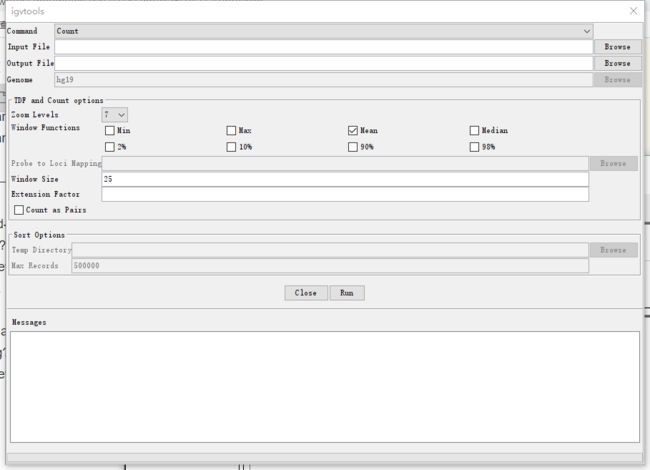
5.3 但是在载入之前需要先将gff3进行排序,选择sort选项