定义
直系同源基因 - Orthologous genes (or orthologs) are a particular class of homologous genes. They are found in different species and have diverged following the speciation of the species hosting them. Therefore, orthologous genes in different species derive from a common ancestral gene found in the ancestor of those species. Given their common origin, it is often the case that orthologous genes have the same function in the different species, but exceptions are not rare. The best way to infer that genes from different species are orthologous is by reconstructing their evolutionary relationships using molecular phylogeny. Conversely, because orthologous genes evolve in parallel with the diversification of species, they are markers of choice for the reconstruction of the evolutionary history of species using molecular phylogeny。
参考连接--Encyclopedia of Astrobiology
基因家族扩张和收缩 - 基因家族(gene family),是来源于同一个祖先,由一个基因通过基因重复而产生两个或更多的拷贝而构成的一组基因,它们在结构和功能上具有明显的相似性,编码相似的蛋白质产物, 同一家族基因可以紧密排列在一起,形成一个基因簇,但多数时候,它们是分散在同一染色体的不同位置,或者存在于不同的染色体上的,各自具有不同的表达调控模式。
在基因组项目中,通常会选择自己要研究的物种和其近缘的物种通过比对来寻找基因家族。通常我们有两种方式来得到基因家族:1 利用orthmcl软件进行blast比对,2将多条物种蛋白muscle比对之后,利用hmmer和pfam数据库比对,如果能比对上同一个蛋白质家族,认为这几条序列是隶属于同一个基因家族。当然也可以两种方法结合起来做。我做项目过程中大多选择第一种,因此今天利用第一种得到的基因家族信息来介绍基因家族收缩和扩张。
参考连接 -- 基因家族收缩和扩张分析 强烈推荐看下。
软件安装
Othofinder:
【Orthofinder】官方文档解读 不建议从conda装
-
服务器安装orthofinder填坑日记
- 注意各个依赖软件的版本,在
config.json中调整相应的命令。比如可以用iqtree调整相应参数达到快速建树的目的: -
"cmd_line": "iqtree -s INPUT -pre PATH/IDENTIFIER -m MFP -bb 1000 -bnni -redo -nt AUTO > /dev/null",可以用iqtree的超快最大似自然法。--IQ-TREE的使用 - 超快速用极大似然法构建进化树
- 注意各个依赖软件的版本,在
Cafe
conda install -c bioconda cafe
r8s
- mac下载Download r8s vers. 1.81解压。
- 直接运行会报错,内容大概是缺少
gfortran - fortran被集成在gcc中,而macOS本身不提供gcc而是clang。所以需要重新安装
gfortran,尝试过用brew安装,好像没效果。 - 去gfortran-github下载gfortran-8.2-Mojave.dmg,然后安装
- linux下解压后make一下,我在服务器上不是root用户,编译起来比较麻烦,也报了很多错,最后放弃了....安装方法参考基因家族扩张与收缩分析及物种进化树构建(上)
seqkit
conda install -c bioconda seqkit -y
直系同源分析
准备工作:
确定需要选择的物种,这些物种在进化上具有层级关系,推荐从文献中获得。
通过查找文献,各种数据库寻找这些物种的相关的基因组测序数据。(genome,cds,pep,gff)
-
不含isoform的蛋白序列文件。
- 原始的pep.fa
- 原始的gff
设置工作目录
使用TBtools制作不含isoform的蛋白质序列文件
网上有各色各样的脚本,但是不同机构提供的gff文件又五花八门,有一定几率出问题。目前我用的最,最方便的肯定是TBtools中的Sequence Toolkit。这里也详细写下方法。比较准确,也没有什么操作上的门槛。
如果在phytozome(需要注册账号)下载蛋白序列,直接下载类似葡萄的Vvinifera_457_v2.1.protein_primaryTranscriptOnly.fa.gz这种包含primaryTranscriptOnly的蛋白序列。
如果是自己测序的或者其他数据库中的蛋白序列,很可能存在可变剪接体的存在比如Gh.A05G000220.1和Gh.A05G000220.2这种,这些isoform会干扰最后的结果,这里用TBtools的sequence toolkits中的一些小工具来提取Primary Transcript
提取代表性转录本ID
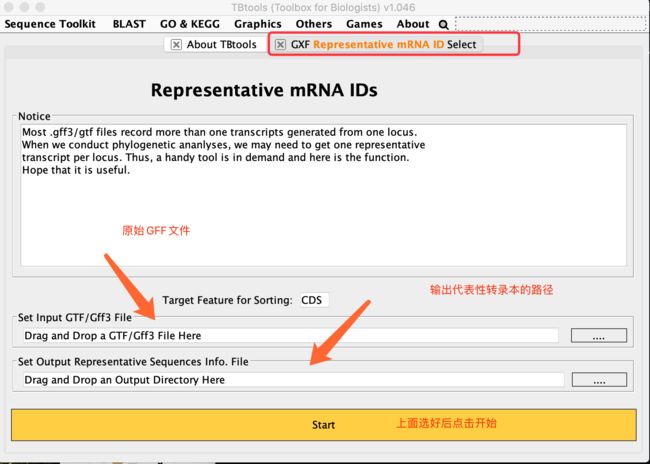
- 把Gff拖入input
- 把你想放输出文件的文件夹拖到output栏里
- 点击start
根据代表转录本ID提取蛋白值序列
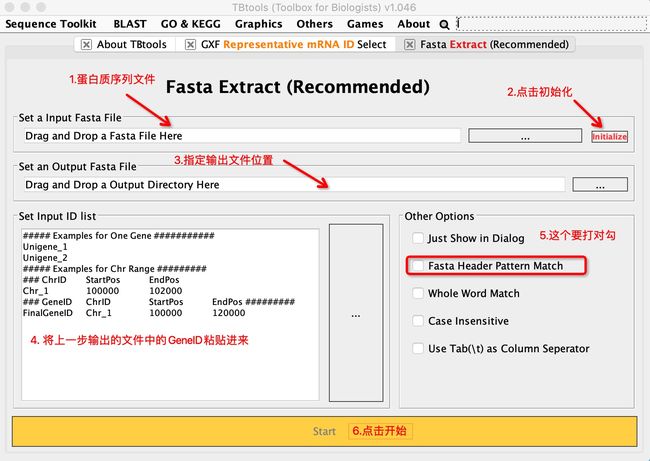
- 必须要初始化。
- fasta header pattern match意思是序列标识,如果看过fasta文件,知道每条序列上面都有类似
> Gh.A01G000020这种,有的人在做序列文件的时候会加上序列的物理位置,注释等,如果有这些东西,需要勾选只匹配header。
orthofinder
将没有isoform的蛋白质序列放在你准备好的工作目录下。
检查下orthofinder以及依赖是否装好了。
一切就绪后运行:
~02.src/07.orthofinder/OrthoFinder/orthofinder -f ~/05.MyPrj/orthofinder/workingdir/ -S diamond -M msa -T raxml -t 8 -a 8
-
-S diamond使用diamond比对速度超级快 -
-M msa基因树推断方法 -
-T raxml这里选择用最大似自然法,也可以选择fasttree, iqtree,raxml-ng -
-t 8 -a 8运行程序需要的核心树,默认单核。
关于系统发育树构建算法问题:在物种发育树的推断上,
OrthoFinder使用STAG算法,利用所有进行构建系统发育树,而非单拷贝基因。此外当使用MSA方法进行系统发育树推断时,OrthoFinder为了保证有足够多的基因(大于100)用于分析,除了使用单拷贝基因外,还会挑选大部分是单拷贝基因的直系同源组。这些直系同源组的基因前后相连,用空缺字符表示缺失的基因,如果某一列存在多余50%的空缺字符,那么该列被剔除。最后基于用户指定的建树软件进行系统发育树构建。结果在WorkingDirectory/SpeciesTree_unrooted.txt使用
STRIDE算法从无根树中推断出有根树, 结果就是SpeciesTree_rooted.txt.来源:徐洲更的第二大脑 -- 使用OrthoFinder进行基因家族分析
更多参数:
OrthoFinder version 2.3.12 Copyright (C) 2014 David Emms
SIMPLE USAGE:
Run full OrthoFinder analysis on FASTA format proteomes in
orthofinder [options] -f
Add new species in to previous run in and run new analysis
orthofinder [options] -f -b
OPTIONS:
-t Number of parallel sequence search threads [Default = 12]
-a Number of parallel analysis threads [Default = 1]
-M Method for gene tree inference. Options 'dendroblast' & 'msa'
[Default = dendroblast]
-S Sequence search program [Default = diamond]
Options: blast, mmseqs, blast_gz, diamond
-A MSA program, requires '-M msa' [Default = mafft]
Options: muscle, mafft
-T Tree inference method, requires '-M msa' [Default = fasttree]
Options: iqtree, raxml-ng, fasttree, raxml
-s User-specified rooted species tree
-I MCL inflation parameter [Default = 1.5]
-x Info for outputting results in OrthoXML format
-p Write the temporary pickle files to
-1 Only perform one-way sequence search
-X Don't add species names to sequence IDs
-n Name to append to the results directory
-o Non-default results directory
-h Print this help text
WORKFLOW STOPPING OPTIONS:
-op Stop after preparing input files for BLAST
-og Stop after inferring orthogroups
-os Stop after writing sequence files for orthogroups
(requires '-M msa')
-oa Stop after inferring alignments for orthogroups
(requires '-M msa')
-ot Stop after inferring gene trees for orthogroups
WORKFLOW RESTART COMMANDS:
-b Start OrthoFinder from pre-computed BLAST results in
-fg Start OrthoFinder from pre-computed orthogroups in
-ft Start OrthoFinder from pre-computed gene trees in
LICENSE:
Distributed under the GNU General Public License (GPLv3). See License.md
CITATION:
When publishing work that uses OrthoFinder please cite:
Emms D.M. & Kelly S. (2019), Genome Biology 20:238
If you use the species tree in your work then please also cite:
Emms D.M. & Kelly S. (2017), MBE 34(12): 3267-3278
Emms D.M. & Kelly S. (2018), bioRxiv https://doi.org/10.1101/267914
如果程序意外断掉了怎么办
之前在做的时候,管理员禁止在主节点跑大程序,所以用qsub投递了任务,但是第一次写pbs脚本的时候没有设置walltime,当时也不理解walltime的意思,最后运行了24个小时程序终止了。当然没有跑完,重新跑一次会花费很长时间,经过github issue板块问作者,以及后来自己看上面的帮助发现作者在软件中设置了重启任务的设置,通过查找pbs输出的日志的脚本发现已经完成了第一步BLAST,所以我第二次在跑任务的时候将代码中加入了-fg 这里的
修改后的pbs脚本:
# !/bin/bash
#PBS -N orthofinder
#PBS -e ~/05.MyPrj/orthofinder/workingdir/tree.error.txt
#PBS -o ~/05.MyPrj/orthofinder/workingdir/tree.out.txt
#PBS -l walltime=240:00:00
source ~/.bashrc
cd ~/05.MyPrj/orthofinder/
~02.src/07.orthofinder/OrthoFinder/orthofinder -fg ~/05.MyPrj/orthofinder/workingdir/Results_Jul02/ -S diamond -M msa -T raxml -t 8 -a 8
我跑了一个14个物种的,可能有亲缘关系较近的原因(考量的生物学问题不同和集群负载的关系所以只能这么做),大概花了两天。
ls -lh
#######################==orthofinder output==###############################################################################
-rw-r--r-- 1 xxx 2.5K Jul 11 23:32 Citation.txt ## 参考文献
drwxr-xr-x 2 xxx 4.0K Jul 11 23:32 Comparative_Genomics_Statistics ## 一些统计信息。
drwxr-xr-x 2 xxx 4.0K Jul 11 23:32 Gene_Duplication_Events ## 基因复制事件
drwxr-xr-x 2 xxx 2.0M Jul 11 23:32 Gene_Trees ## 基因家族进化树
-rw-r--r-- 1 xxx 956 Jul 11 23:32 Log.txt ## 工作日志记录
drwxr-xr-x 2 xxx 2.0M Jul 11 23:16 MultipleSequenceAlignments ## 基因家族的多序列比对,用于上面基因家族进化树构建
drwxr-xr-x 2 xxx 4.0K Jul 10 22:41 Orthogroups ## 同源基因组*下面有文件结构
drwxr-xr-x 2 xxx 3.5M Jul 10 22:41 Orthogroup_Sequences ## 各个基因家族成员序列
drwxr-xr-x 16 xxx 4.0K Jul 11 23:17 Orthologues ## 直系同源关系
drwxr-xr-x 2 xxx 4.0K Jul 11 23:18 Phylogenetically_Misplaced_Genes ## 系统演化上错位的基因*字面意思,需要后续看文献理解算法
drwxr-xr-x 2 xxx 4.0K Jul 11 23:17 Putative_Xenologs ## 可能的异同源基因
drwxr-xr-x 2 xxx 2.0M Jul 11 23:32 Resolved_Gene_Trees ## ...后续看文献
drwxr-xr-x 2 xxx 256K Jul 10 22:42 Single_Copy_Orthologue_Sequences ## 单拷贝基因序列
drwxr-xr-x 2 xxx 4.0K Jul 11 23:17 Species_Tree ## 构建的物种树为STAG树
drwxr-xr-x 6 xxx 32K Jul 28 20:03 WorkingDirectory ## 工作目录
#######################==orthofinder output==###############################################################################
tree Orthogroups
Orthogroups/
├── Orthogroups.GeneCount.tsv ## 每个家族在每个物种物种中的成员数量
├── Orthogroups_SingleCopyOrthologues.txt ## 单拷贝基因
├── Orthogroups.tsv ## 每个物种各个家族成员的表格
├── Orthogroups.txt ## 每个家族所有成员(物种混合在一起)
└── Orthogroups_UnassignedGenes.tsv ## 直译过来未指派的基因,可以物种独有的
## 那些包含序列,或者家族树的路径下有上万个文件,ls的话会卡很久,如果想看几个可以使用
ls | head -10 # 将ls的结果管道传递给head 用-num来调整你想看前几个或者 tail看最后几个
- 在
Orthogroups里面可以找到基因家族成员统计,这里的Orthogroups.GeneCount.tsv会在后续的cafe中用到。 -
Orthogroups.txt Orthogroups.txt做基因家族的同学可能会感兴趣,用excel或这grep等命令搜索下你做的成员,看这些成员在哪个orthogroup中,这个orthogroup应该就是你要找的家族,至少是个亚家族。可以和之前用隐马模型或者blast找到的家族成员结果验证下。 - 在
Gene_Trees下面有每个家族的进化树,如果确定了你研究的家族可以将这个树导出来看下家族成员的系统发育。 - 在
Species_Tree下有经过最大似自然法构建的STAG物种树。 - 在
Orthologues路径下有物种之间互相比较的直系同源关系,可以根据这种直系同源关系计算两个物种间的进化选择压力(ka/ks)进行分子进化研究。物种间的ks的分布统计结合synonymous substitutions per synonymous site per year (r)值通过下面公式统计分化时间
其他分析可以结合文献挖掘有用的信息。这里感叹下作者的牛逼。
基因家族的扩张和收缩分析
用conda装好cafe后发现还需要一些脚本,可以从这里下载cafe tutorial, 可能需要访问外网,我把其中的python脚本做了备份放在我gitee的一个常用代码仓库的python下:MyScript-gitee 仓库有点乱,没有整理,需要的脚本在python/01.cafe中。同时我把一个cafe结果可视化的脚本CAFE_fig.py也放到里面了。
r8s构建超度量树
超度量树(ultrametric tree)也叫时间树,就是将系统发育树的标度改成时间,从根到所有物种的距离都相同。构建方法有很多,比较常用的就是 r8s . --「基因组学」使用CAFE进行基因家族扩张收缩分析
Ultrametric Tree An ultrametric tree is a rooted tree with edge lengths where all leaves are equidistant from the root. Often, ultrametric trees represent the molecular clock which states that the rate of mutation is the same across all lineages of the tree
超度量树的计算方法和构建过程:Phylogenetic Trees and Multiple Alignments
准备r8s输入文件
这里需要使用cafetutorial_prep_r8s.py脚本
- orthofinder结果中
Species_Tree下的SpeciesTree_rooted.txt文件,根据这个物种树来构建超度量树。 - 去timetree网站在你输入物种中找两个已经预测过分化年限的物种,输入拉丁文名,网站会给你一个分化年限,这里要作为r8s的一个参数。
- 在
MultipleSequenceAlignments下有构建物种树的序列比对文件SpeciesTreeAlignment.fa,进来后首先用seqkit来提取用于物种树序列比对的aa数目这里特别感谢 CoCo正能量 大神的指导❤️ - 参考文章:基因家族扩张与收缩分析及物种进化树构建(上)
## seqkit提取物种树比对的氨基酸序列数目
seqkit stat MultipleSequenceAlignments/SpeciesTreeAlignment.fa
file format type num_seqs sum_len min_len avg_len max_len
MultipleSequenceAlignments/SpeciesTreeAlignment.fa FASTA Protein 14 31,629,122 2,259,223 2,259,223 2,259,223
## 看下脚本参数
python ../01.script/cafetutorial_prep_r8s.py --help
#######################==脚本参数==###############################################################################
optional arguments:
-h, --help show this help message and exit
-i INPUT_FILE, --input-file INPUT_FILE
full path to .txt file containing tree in NEWICK ## 树文件NEWICK格式
format
-o OUTPUT_FILE, --output-file OUTPUT_FILE
full path to file to be written (r8s input file) ## 输出文件位置
-s SITES_N, --sites-n SITES_N
number of sites in alignment used to estimate species ## 序列比对文件中序列长度
tree
-p SPP_PAIRS, --pairs-species SPP_PAIRS ## 哪一对物种作为校准时间点物种(拉丁文名字)
-c CAL_POINTS, --calibration-points CAL_POINTS ## 校准时间点(MYA)
#######################==脚本参数==###############################################################################
## 执行脚本,注意这里校准物种的编号要和你放在工作目录中蛋白序列的名字一致
python ../01.script/cafetutorial_prep_r8s.py -i SpeciesTree_rooted.txt -o ../03.r8s_file/r8s_ctl_file.txt -s 2259223 -p 'B.ceiba,G.raimondii' -c '33'
## 查看准备好的r8s准备文件
cat r8s_ctl_file.txt
#######################==r8s准备文件内容==###############################################################################
#NEXUS
begin trees;
tree nj_tree = [&R] <这里是物种树的文件>
End;
begin rates;
blformat nsites=2259223 lengths=persite ultrametric=no; # 这里是你刚才输入的nsites
collapse;
mrca ibadii B.ceiba G.raimondii; # 这里是校准物种
fixage taxon=ibadii age=24; # 注意这个ibadii,是你校准物种后3个字符合起来的,标记为校准分叉节点。
divtime method=pl algorithm=tn cvStart=0 cvInc=0.5 cvNum=8 crossv=yes;
describe plot=chronogram;
describe plot=tree_description;
#######################==r8s准备文件内容==###############################################################################
r8s计算超度量树
## 运行r8s
~/01.src/r8s1.81/src/r8s -b -f r8s_ctl_file.txt >r8s_tmp.txt
## 打开这个结果文件可以发现在文件的末尾有一个树,这个树就是我们要的超度量树,可以通过上面提到文章中提供的代码把树提取出来
tail -n 1 r8s_tmp.txt | cut -c 16- > twelve_spp_r8s_ultrametric.txt
这里需要注意我们对构建的超度量树需要去掉校准物种节点,不然cafe会报错,树文件缺少时间。
((A:0.596980,B:0.534235):0.534121,(C:0.23123,D:0.23314):0.234441,E:0.123212)ibadii; ## 随便瞎写的树,最后那个ibadii就是我说的节点标识符,需要手动去掉,去掉后的树就是
((A:0.596980,B:0.534235):0.534121,(C:0.23123,D:0.23314):0.234441,E:0.123212)
运行cafe
具体参考 使用OrthoFinder进行基因家族分析 基因家族扩张与收缩分析及物种进化树构建(上)
这里需要把我们orthofinder结果中Orthogroups.GeneCount.tsv改造成cafe需要的格式。首先看下cafe需要的文件格式
##===================Cafe 要求的文件格式===================##
Desc sp-A sp-B sp-C sp-D sp-E sp-F sp-G sp-H sp-I sp-J sp-K sp-L sp-M
(null) OG0000000 3 37 12 24 39 51 31 4 3 73 95 15 62 69
(null) OG0000001 8 23 21 27 32 42 1 26 30 19 48 5 34 36
(null) OG0000002 18 21 15 19 23 29 6 27 30 22 24 37 26 42
##===================Cafe 要求的文件格式===================##
再看下Orthogroups.GeneCount.tsv
##===================Orthogroups.GeneCount.tsv===================##
Orthogroup sp-A sp-B sp-C sp-D sp-E sp-F sp-G sp-H sp-I sp-J sp-K sp-L sp-M Total
OG0000000 3 37 12 24 39 51 31 4 3 73 95 15 62 69 518
OG0000001 8 23 21 27 32 42 1 26 30 19 48 5 34 36 352
OG0000002 18 21 15 19 23 29 6 27 30 22 24 37 26 42 339
##===================Orthogroups.GeneCount.tsv===================##
需要做的是:
- 去掉total这一列
- 在第一列添加Desc,然后这一列都为(null)
# 为了方便(偷懒)用&&写成了一行
awk 'OFS="\t" {$NF=""; print}' Orthogroups.GeneCount.tsv > tmp && awk '{print "(null)""\t"$0}' tmp > cafe.input.tsv && sed -i '1s/(null)/Desc/g' cafe.input.tsv && rm tmp
- 这里指定
OFS="\t"即输出文件的分隔符是制表符 -
$NF=""就是最后一列为空 - 第二段在整个文件前面加一列为
(null)的列 - 第三段
sed -i在原文件中修改 -
'1s/(null)/Desc/g'1s指定第一列,将第一列中的(null)替换成Desc - 最后删掉中间文件
对文进行过滤
第二步,将那些基因拷贝数变异特别大的基因家族剔除掉,因为它会造成参数预测出错。下面的脚本是过滤掉一个或多个物种有超过100个基因拷贝的基因家族,虽然不是特别的严格,但效果和根据拷贝数变异过滤类似。
python ../01.Script/cafetutorial_clade_and_size_filter.py -i cafe.input.tsv -o filtered.cafe.input.tsv -s 2> filtered.log
cafe.run配置文件
#!~/miniconda2/envs/cafe/bin/cafe
load -i filtered.cafe.input.tsv -t 4 -l reports/log_run1.txt -p 0.05
tree ((A:0.596980,B:0.534235):0.534121,(C:0.23123,D:0.23314):0.234441,E:0.123212)
lambda -s -t ((1,1)1,(1,1):1,1)
report reports/report_run1
- 第一部分
load后面跟刚才准备好的文件。 - 第二部分为之前做好的r8s构建的时间树。
-
lambda,如果你认为这些物种间分子进化速率不同,可以按照物种分开比如E和ABC的进化速率不同,lambda -s -t ((1,1)1,(1,1):2,2) -
report输出文件位置
都准备好后执行
cafe cafe_run1.sh
Cafe结果处理
对于结果文件的解读不在造轮子,请看:「基因组学」使用CAFE进行基因家族扩张收缩分析
没有继续研究之前说过的python脚本,后续应该又很多可以拓展的,时间关系,只是follow了几位大佬的过程。
用cafetutorial_report_analysis.py导出相应比较好读的文件:
# 执行脚本
python cafetutorial_report_analysis.py -i reports/report_run.cafe -o reports/summary_run
## 结果文件结构
├── log_run1.txt # 输出文件结果日志
├── report_run1.cafe # 输出文件
├── summary1_anc.txt # 包括每个节点,每个branch上面家族成员的数量表格
├── summary1_fams.txt # 每个节点,每个branch中家族具体成员
├── summary1_node.txt # 每个节点,每个branch中基因家族扩张和收缩的数目
└── summary1_pub.txt # 更加具体的数量统计表,包括扩张/收缩基因比例等等
##=============== summary1_anc.txt 示例 ====================##
Family ID <11> A<16> <21> B<4> <23> C<22> D<18> E<20> F<2> G<14> H<24> ...
OG0000000 0 39 0 24 0 62 12 4 69 37 73 3 ...
OG0000001 0 32 0 27 0 34 21 26 36 23 19 8 ...
OG0000002 0 23 0 19 0 26 15 27 42 21 22 18 ...
##=============== summary1_anc.txt 示例 ====================##
################################################################################
##=============== summary1_fams.txt 示例 ====================##
# The labeled CAFE tree: ((((((A<0>,B<2>)<1>,(C<4>,D<6>)<5>)<3>,E<8>)<7>,...
Overall rapid : OG0000000,OG0000001,OG0000002,OG0000003,OG0000004,OG0000005,OG0000006,OG0000007,OG0000008,OG0000009,OG0000010,OG0000011,OG0000012,OG0000013,OG0000014,OG0000015,OG0000016,OG0000017,OG0000018,OG00000
A<6>: OG0000000[+51*],OG0000001[+42*],OG0000002[+29*],OG0000003[+2*],OG0000004[+21*],OG0000005[+27*],OG0000006[+22*],OG0000007[+23*],OG0000008[+46*],OG0000009[+19*],OG0000010[+25*],OG0000011[+14*],OG0000
GB<10>: OG0000000[+15*],OG0000001[+5*],OG0000002[+37*],OG0000003[+2*],OG0000004[+19*],OG0000005[+22*],OG0000006[+27*],OG0000007[+24*],OG0000008[+27*],OG0000009[+18*],OG0000010[+16*],OG0000011[+12*]
##=============== summary1_fams.txt 示例 ====================##
################################################################################
##=============== summary1_node.txt 示例 ====================##
Node Expansions Contractions Rapidly evolving families
A<6> 5300 1207 6507
B<10> 4400 1073 3650
<21> 0 0 0
##=============== summary1_node.txt 示例 ====================##
################################################################################
##=============== summary1_pub.txt 示例 ====================##
Species Expanded fams Genes gained genes/expansion Contracted fams Genes lost genes/contraction No change Avg. Expansion
A 5300 (5300) 8735 1.65 1207 (1207) 1438 1.19 31857 0.190204
B 4400 (3458) 6765 1.54 1073 (192) 1361 1.27 32891 0.140861
##=============== summary1_pub.txt 示例 ====================##
有了扩张和收缩的数目,就可以根据这些数据来进行后续的分析,而且还包括了每个家族的扩张和收缩具体信息。
图形话展示的话,也可以通过itol,evolview等工具自行添加信息。比如pie图,数值等等,后续在AI里编辑。
==end==