predicting Growth and Carcass traits in swine Using Microbiome Data and Machine Learning Algorithms
使用微生物组数据和机器学习算法预测猪的生长和胴体性状
Christian Maltecca,Due Lu
阅读者:王靖文
摘要:在本文中,我们评估了在生长测试期(断奶期,15周和22周)的三个时间点采取的微生物组测量的功效,以预测一系列杂交猪的1039个个体的生长和胴体性状。我们将预测准确度测量为五重交叉验证设置中实际和预测表型之间的相关性。测量的表型性状包括活体重量测量和在试验期间以及屠宰时获得的胴体组成。我们采用了一个零模型,不包括微生物组信息作为基线来评估预测准确性的增加,这是因为将操作分类单位(OTU)作为预测因子。我们进一步对比了贝叶斯套索(Bayesian Lasso)以及机器学习方法(随机森林和梯度提升)和半参数核心模型(再生核心希尔伯特空间)的模型的性能。在大多数情况下,预测精度随着微生物群数据的加入而显著提高。在第15周和第22周采集的微生物群信息,其准确性更为显著,其值从腰状特征的0.30到背部脂肪的0.50以上不等。相反,断奶时的微生物组组成导致大多数情况下预测准确性的边际增益,这表明后期测量可能更有助于在预测模型中纳入。模型选择对预测的影响很小,没有任何模型/特征/时间点的明显赢家。因此,我们建议将跨模型的平均预测作为拟合微生物组信息的稳健策略。总之,微生物组组合物可以有效地用作生长和组成性状的预测因子,特别是对于肥胖性状。加入otU预测因子可能会促进个体的快速生长,同时限制脂肪积累。早期的微生物群落测量可能不是生长的良好预测指标,otU 信息最好在后期生命阶段收集。未来的研究应侧重于将微生物组和宿主基因组信息纳入预测,以及两者之间的相互作用。此外,应研究微生物组对饲料效率以及胴体和肉质的影响。
生产可销售肉类产品的效率主要取决于与饲料相关的成本以及所生产的瘦肉的数量和质量。更有效地利用饲料资源已成为畜牧业面临的一项挑战。最近一直在努力发现和利用个体猪的基因组变异性,提高饲料效率。尽管取得了成功,但这种方法还存在与获得准确的个体采食记录以及定义和使用不同饲料效率措施相关的后勤和技术限制。也许最重要的是,持续的努力只集中在猪的效率变异上,这将不可避免地导致边际收益减少,随着时间的推移伴随着整体适应性和遗传多样性的丧失。存在于个体肠道中的细菌的数量和类型是所有哺乳动物的关键部分。微生物组的构成代表了巨大的基因组的多样性,其有助于生理和健康。特别是,肠道微生物组直接影响碳水化合物的降解,提供短链脂肪酸,减轻和改变潜在有毒化合物的影响,并产生必需的维生素。环境因素的影响,如营养压力因素,以及与断奶和管理相关的挑战在猪身上已经表征出来。尽管如此,健康的微生物生态系统的组成和功能尚未被定性和定量地定义,并没有被用作最大限度提高动物健康和性能的工具。特别是,微生物组组成尚未在大规模上进行研究,包括通过几个生产阶段进行的大量抽样。在本文中,我们评估了基于粪便样本的微生物组预测的能力,以预测健康杂交猪群体的生长和胴体组成。在这样做的过程中,我们采用了典型的宿主基因组预测机制,包括贝叶斯字母模型以及半参数和机器学习算法。
结果
在这项工作中,我们评估了纵向微生物组数据的有效性,以预测猪的生长和胴体组成。 为此,我们使用并对比模型,在基因组选择领域被证明是成功的,以便为将来在选择计划中将微生物群信息纳入日常目的提供蓝图。我们在交叉验证设置中评估了所提出的模型的性能。我们进一步测试了基于混合模型的后分析的整体实验设计。
随着时间的推移而改变的微生物群。 Lu及其同事最近详细描述了当前数据中三个时间点(断奶,15周和22周)的生物分类丰度分布。由于当前论文的目的不是提供所测数据的生态景观,因此请读者参考该论文以获取更多详细信息。简而言之,在养猪的三个不同阶段,分别有14、21、29、54、106和202个已识别的门、纲、目、科、属、种。对于这三个采样点,将95.79-97.80%的OTU分为六个门:硬毛菌,拟杆菌属,变形杆菌,融合细菌,螺旋体和放线菌。菌门中的细菌占总种群的大部分,其次是拟杆菌。为了评估微生物组预测表型指标的能力,我们进行了初步分析,以调查不同的采样时间如何影响粪便的微生物组的组成。为此,我们拟合了一个随机森林模型,该模型与用于生长和胴体特征的模型相似(请参见方法),在这种情况下唯一的区别是,该模型用于将每个观测值划分为三个采样时间之一。我们在图1中报告了五重分类的结果,该结果描述了在断奶,15周和22周时的归一化分类混淆矩阵。单个时间测量构成了三个不同的微生物种群。在所有情况下,分类的准确性都非常高(> 95%)。 15周和22周的误分类率略高(-3%)。这一结果与Lu及其同事的报告相一致,在断奶时发现了两种不同的微生物肠型,但在后来的时间点发现了两种不同的聚集型。在补充材料中可以找到其他信息,其中报告了随时间变化的丰度(补充图1),主坐标分析(补充图2)和不同时间点种群的显著对数倍数变化(补充图3)
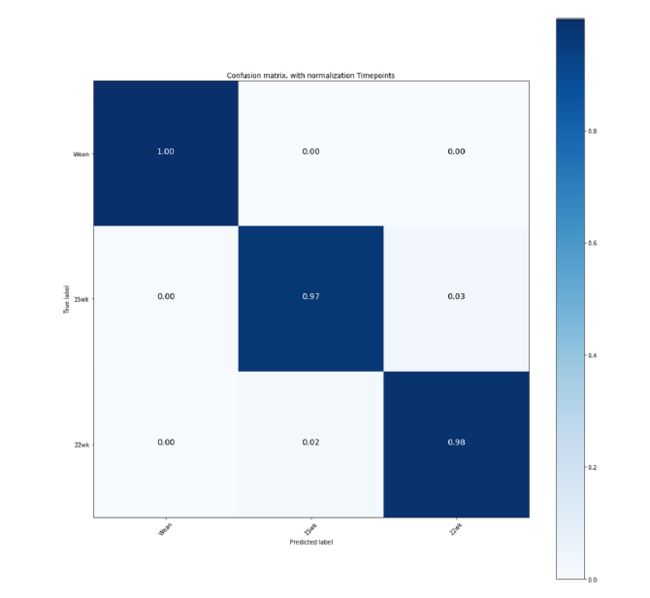
图1. 三个时间点的微生物群落成分的标准化分类混淆矩阵。Wean=断奶15wk = 15 weeks, 22wk = 22 weeks. 通过 RF 模型从五倍交叉验证中获得的混淆矩阵。
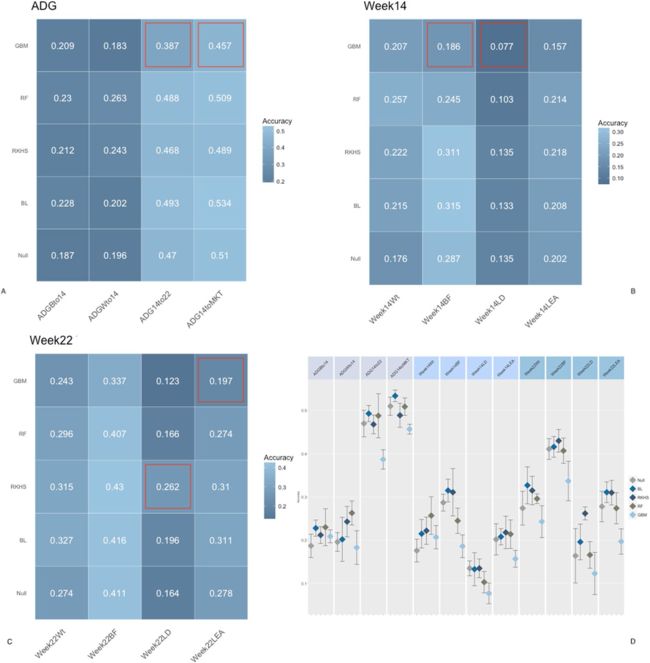
图2.断奶时微生物组组成预测的准确性。组(A)日增重特征的准确性,组(B)14周特征的准确性,组(C)22周特征的准确性,组(D)模型/特征组合的90%置信区间。 使用RF模型从五重交叉验证中获得的混淆矩阵。 BL =贝叶斯套索,RF =随机森林,GBM =梯度提升机,RKHS =复制内核希尔伯特空间。ADGBto14 =到第14周的平均日增重,ADGWto14 =到第14周的平均日增重,ADG14to22 =第22到第22周的日均增重,ADG14toMKT =进入市场的第14周的平均日增重, Week14Wt = 第14周的体重, Week14BF = 第14周的背脂, Week14LD = 第14周的腰部深度, Week14LEA =第14周的腰部眼肌, Week22Wt = 第22周的体重, Week22BF =第22周的背脂, Week22LD = 第22周的腰部深度, Week22LEA =第22周的腰部眼肌. 红色轮廓线表示预测与零模型有显著差异。
图3.第15周微生物组组成预测的准确性。图(A)为日增重性状的准确性,图(B)为第14周性状的准确性,图(C)为第22周性状的准确性,图(D)90%置信度 模型/特征组合的间隔。 使用RF模型从五重交叉验证中获得的混淆矩阵。 BL =贝叶斯套索,RF =随机森林,GBM =梯度提升机,RKHS =复制内核希尔伯特空间。 ADGBto14 =到第14周的平均日增重,ADGWto14 =到第14周的平均日增重,ADG14to22 =第22周到第22周的平均日增重,ADG14toMKT =进入市场的第14周的平均日增重,Week14Wt = 第14周的体重, Week14BF= 第14周的背脂, Week14LD= 第14周的腰部深度, Week14LEA= 第14周的腰部眼肌, Week22Wt= 第22周的体重, Week22BF= 第22周的背脂, Week22LD= 第22周的腰部深度, Week22LEA=第14周的腰部眼肌. 红色轮廓线表示预测与零模型有显著差异。
交叉验证突出了微生物组对生长和胴体预测的显著作用。
我们首先评估了微生物组数据在预测健康杂种公羊中若干生长参数方面的功效,这些杂种公羊起源于28个成年父系的交配。为此,我们考虑了:在生长试验的第14周和第22周测量的体重,背部脂肪,腰部面积和深度特征以及同一时期的日增重量。这些与在断奶时以及试验的第15周和第22周从同一个人获得的粪便微生物组信息结合在一起。使用交叉验证方案对每个特征进行独立分析,其中使用一些样本的表型和OTU训练统计模型,其余用于验证预测。我们在分析中考虑了三类模型:一种来自贝叶斯字母系的模型,贝叶斯拉索(BL);两种机器学习方法,随机林(RF)和梯度提升机(GBM);和一个半参数方法,再生内核希尔伯特空间(RKHS)。我们选择这些模型作为牲畜和农作物中基因组预测最广泛使用的方法的代表。我们这样做是为了强调当前工作中提出的分析与基因组选择方法在范围和方法上的相似性,并提供了扩展的基础,将基因组信息纳入未来的比较。
图2、3和4报告了每种性状,粪便微生物组时间点和方法组合的预测准确性。微生物组对预测的贡献被测量为与零模型的偏差,该模型仅包括性别,父系,断奶时体重和重复的影响。应该注意的是,在所有情况下,在提出的每种算法中都拟合了空模型。为了便于比较,将空模型的性能表示为各个方法中空模型的平均值。在大多数情况下,相对于零模型,将OTU丰度包含在预测模型中可提高准确性。尽管如此,该量根据微生物组时间点而变化。通常,在断奶时包括微生物组组成对于日增重性状以及在第15周和第22周获得的胴体指标具有较低的预测能力(图2)。对于日增重特征(面板A),微生物组信息的纳入使预测准确性提高了约3%,但在所有情况下,对于所有算法,预测的90%CI(面板C)在null模型和biom模型之间重叠采用。无论微生物组信息是否包含在内,都比早期生长更好地预测了试验后期的日增重。在第14周和第22周测得的胴体性状也观察到类似的趋势,对于零模型和OTU模型,预测范围从腰部深度的约15%(B,C组)到背脂肪的约40%。相反,第15周的微生物组组成大大提高了测试集中的准确性(图3)。数量取决于性状/时间组合。总的来说,正如预期的那样,对于与微生物组采样同时进行测量的性状,微生物组组成的预测准确性更高。对于日常增益特征(面板 A),包含微生物群信息可提高早期生长预测的准确性,从出生到第 14 周和从绝育到第 14 周每天增益的空模型的预测精度为 ±20%,对相同的两个特征而言,从 14 到 40 和 45%。同样,对于在第14周测量的所有特征(图B),微生物组信息显著提高了预测准确性,体重和背脂肪的增幅分别为~0.20,腰部深度和面积的增幅分别为~0.05和0.10。第22周的性状也有相似的趋势,尽管增加的幅度较小,并且某些性状(体重除外)重叠90%CI(图D)。图4描绘了在第22周测量的微生物组的交叉验证预测结果。应当指出,鉴于采样的时间连续,由于措施的时间继承,在第14周测量的表型和第22周的微生物群的组合应谨慎解释。同样,对于大多数性状,微生物组信息提高了预测准确性。但是,对于大多数特征/模型组合而言,增加并不显著。具体来说,着眼于第22周的性状,只有OTU才使体重和背脂受益,背脂的增幅为~0.08,体重的增幅为~0.05。有趣的是,包括OTU的丰度并没有提高以后日收益特征(从第14周到第22周以及从第14周到上市)的预测准确性。
给出的结果与其他研究中观察到的结果一致。他和他的同事发现,猪肠道微生物组对脂肪有中等程度的影响,微生物组的平均表背脂肪和腹部脂肪重量的表型差异分别为1.5%至2.73%。同样,Fang及其同事在生长中的猪中发现了119个与肌肉内脂肪相关的OTU。此外,McCormack等鉴定了几种可能与猪饲料效率有关的肠道微生物,Yang等鉴定了杜洛克猪中与残留饲料摄入有关的两种潜在肠型。关于日增重和体重的数据还比较稀少,例如,Ramayo等。根据OTU的丰度确定了仔猪群,这些猪群与60天体重和平均日增重显著相关。值得注意的是,在大多数情况下,这些研究都侧重于细菌生态种群的识别或与特定表型相关的特定OTU的识别。就我们所知,这是首次尝试严格表征微生物组对猪和一般牲畜的生长和胴体性状的总体预测能力。在我们的分析中,大多数情况下,微生物组组成数据的加入提高了预测准确性,超出了确定几个重要生物分类单位的预期精度,与几种牲畜物种的基因组预测中观察到的结果无异,表明不同OTU和微生物群组合物之间的互连比先前研究中强调的更为复杂。此外,越来越多的文献表明猪和它的元基因组之间有着丰富的相互作用。有效地将微生物组信息纳入选择程序既是挑战,也是机遇。微生物群可能被认为是一个完全环境变异的来源,但至少部分是处于宿主的直接控制之下。当前分析中使用的方法在整合由微生物组和宿主基因组数据的可用性所产生的全范围变异性方面将被证明具有极大的灵活性。这些方法中的某些方法可以直接按照GxE示例应用于植物和牲畜。
选择模型会部分影响预测准确性,其结果取决于时间特征组合。我们调查了将微生物组信息纳入猪生长和胴体表型预测的不同模型类别的有效性。我们选择了从完全参数,半参数到非参数的模型,以识别并且可能捕获OTU组成的复杂的相互依存结构。针对每个特征时间点组合对模型进行了独立测试。我们将包括微生物群成分在内的模型与仅包括一般设计因子的基线模型进行比较,从而评估性能(参见方法)。贝叶斯套索是“贝叶斯字母” 家族的模型之一,由于其能够有效处理基因组预测中的小问题,并提供了特征选择的框架,因此在基因组选择中广受欢迎。 BL由Xu等人和de los Campos等人提出。我们选择它作为参数模型类别中最强大和最受欢迎的选择之一。再现内核希尔伯特空间是一类特别灵活的半参数模型,已被提出来拟合复杂的多维数据。由于Gianola和同事和de los Campos等人的工作,它们最近在牲畜和农作物育种中越来越受欢迎。此类模型依赖于选择适当的内核,然后用于与育种设置中常用的混合模型不同的形式模型。随机森林是一种将决策树拟合到数据集的各种子样本的整体方法。随机林模型通常具有强于过度拟合性,可以捕获数据中的复杂交互结构。梯度提升是一种替代性的集成方法,旨在通过形成具有比单个预测器更高的预测能力的预测器委员会,在这种情况下以顺序方式组合预测器。
图3、4和5的面板D描绘了每个模型特征组合的点估计和90%CI。在大多数情况下,模型的选择是洗。在我们的分析中,我们无法确定一个明确的赢家,并且大多数情况下,模型的配置项存在很大重叠。就CI的排名和大小而言,可再生内核希尔伯特空间模型成为最稳定的方案,其次是贝叶斯套索和随机森林,而梯度提升在整个特征时间表现出最大的变化。在断奶时,梯度提升模型在某些情况下的表现要比零模型差。但是,这并不奇怪,因为在大多数情况下,断奶时的微生物组数据对模型的学习几乎没有贡献。我们的结果与在植物和家畜中通过基因组信息预测复杂性状所观察到的结果相似,在不同条件下,不同类别的模型表现相似,因此在大多数情况下,模型的选择是比潜在的生物信号更依赖数量和数据结构。需要注意的是,虽然DNA多态性-信息预测标记信息在某种程度上是一个固定参数,OTU组合体在个体和实验设置之间可能更具变异性,由于取样程序、环境条件以及用于获取分类单元的生物信息学机械的变化。虽然我们确实认识到,某些变异性无法通过统计建模得到有效管理,但我们也认为,其中一些模型在处理此类变异源时可能更为灵活。这应该是进一步研究的主题,并且超出了本文的范围。
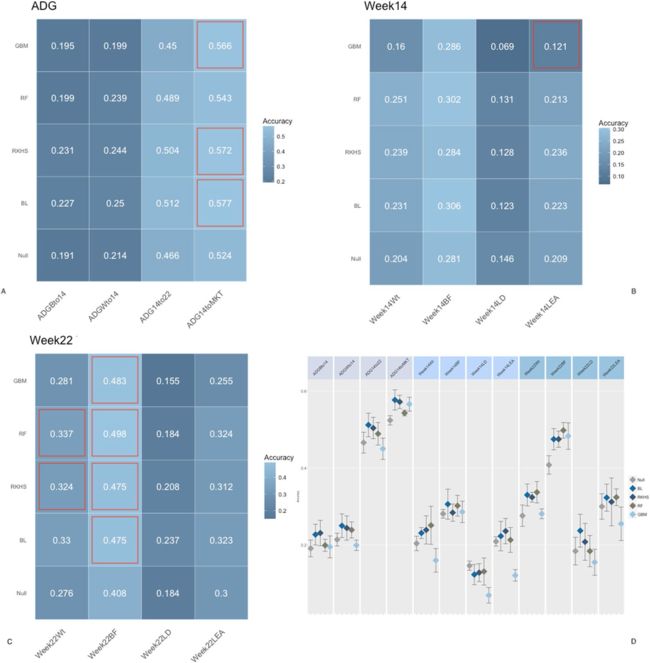
图4.第22周时微生物组组成的预测准确性。图(A)为日增重性状的准确性,图(B)为第14周性状的准确性,图(C)为第22周性状的准确性,图(D)90%置信度 模型/特征组合的间隔。 使用RF模型从五重交叉验证中获得的混淆矩阵。 BL =贝叶斯套索,RF =随机森林,GBM =梯度提升机,RKHS =复制内核希尔伯特空间。 ADGBto14 =到第14周的平均日增重,ADGWto14 =到第14周的平均日增重,ADG14to22 =第22周到第22周的平均日增重,ADG14toMKT =进入市场的第14周的平均日增重,Week14Wt = 第14周的体重, Week14BF = 第14周的背脂, Week14LD = 第14周的腰部深度, Week14LEA = 第14周的腰部眼肌, Week22Wt = 第22周的体重, Week22BF = 第22周的背脂, Week22LD = 第22周的腰部深度, Week22LEA =第14周的腰部眼肌. 红色轮廓线表示预测与零模型有显著差异。
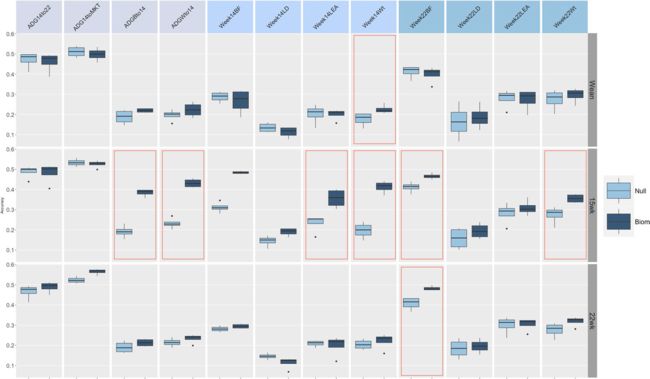
图5.断奶第14周和第22周时模型预测微生物组组成的平均准确度。Null =空模型的平均值。 生物群=微生物组模型的平均值。 ADGBto14 =到第14周的平均日增重,ADGWto14 =到第14周的平均日增重,ADG14to22 =第22周到第22周的平均日增重,ADG14toMKT =进入市场的第14周的平均日增重,Week14Wt = 第14周的体重, Week14BF = 第14周的背脂, Week14LD = 第14周的腰部深度, Week14LEA = 第14周的腰部眼肌, Week22Wt = 第22周的体重, Week22BF = 第22周的背脂, Week22LD = 第22周的腰部深度, Week22LEA =第14周的腰部眼肌. 红色轮廓线表示预测与零模型有显著差异。
在这项工作中,并认识到这种复杂性,我们试图克服这些限制,获得预测精度平均模型。该分析的结果是通过汇总重复项和方法中的信息而获得的,并显示在图6中。在这种情况下,结果提供了两个相互竞争的模型:一个无效模型(再次通过各种方法合并了无效拟合)和一个微生物组模型(biom),通过平均每个特征/方法组合的效果来获得。大部分结果概括了上一节中的内容。在某些情况下,无效模型和微生物组模型之间的差异缩小了(例如,第22周的背部脂肪)。表1中报告了竞争性状/模型组合的均方误差(MSE)。结果大部分概括了MSE的精度,对于包括微生物群信息在内的模型,尤其是wk15和wk22,以及随特征/时间点而变化的模型。不过,与零模型相比,大多数情况下的差异更细微,在某些情况下(例如Week14Wt和Week22Wt),微生物组模型在MSE方面的表现不如零模型。因此,应谨慎解释某些比较的结果,并应进行更大样本量的进一步研究。
后分析结果。我们试图通过交叉验证研究的后分析来评估设计中所有因素对预测性能的总体影响。为此,我们采用了标准的LMM方法(请参见方法),获得分析中所有变量的最小平方平均估计值和对比度。即我们拟合了包含微生物群信息的效果、用于分析的算法、粪便微生物群采样的时间点、分析的特征以及所有成对相互作用。在这种情况下,响应变量是交叉验证实验中预测的准确性。该研究的结果记录在表2和图7中。表2报告了整个实验设计的III型ANOVA。除了算法和特质之间的相互作用外,所有因素及其相互作用都非常重要。算法和时间点之间的交互作用也刚好低于P <0.05的显著性阈值。图7描绘了主要主效应及其相互作用的最小二乘法。微生物组数据的纳入(所有其他因素的平均值)使模型的预测能力比无效模型提高了约4%(0.321对0.281)。在前面提到的模型中,GBM是预测能力最低的模型(0.26),而RKHS是预测能力最高的模型(0.32),尽管与贝叶斯拉索算法和随机森林算法几乎相同。与断奶相比,第15周收集的微生物组信息具有最高的预测能力(0.335),而断奶的前两者之间的差异最低。分别为〜5%和〜4%。每日增益特征和背部脂肪特征预测最好,而腰部特征,无论是面积还是深度,其准确性最低。不同模型之间的相互作用以及微生物组数据的包含再次表明,无论是否存在微生物组数据,RKHS模型均表现最佳。有趣的是,随机森林算法和梯度增强算法都是通过包含OTU信息获得最大收益的算法,与两种情况下的5%的零模型相比都有改进。时间点-算法交互方面也观察到了类似的趋势。最后,微生物群信息与时间点的相互作用突出表明,在我们的数据中,第 15 周收集的微生物群信息的表现大为优于 (约10%)所有其他时间点(以及无效模型)。据我们所知,这是正式评估牲畜中微生物组预测的首次尝试。可比模型已与人类微生物组数据一起用于预测疾病,并与土壤微生物组数据一起用于预测作物产量。在这两种情况下,微生物组数据的使用都提高了预测能力,但是鉴于范围和措施的多样性,很难进行直接比较。
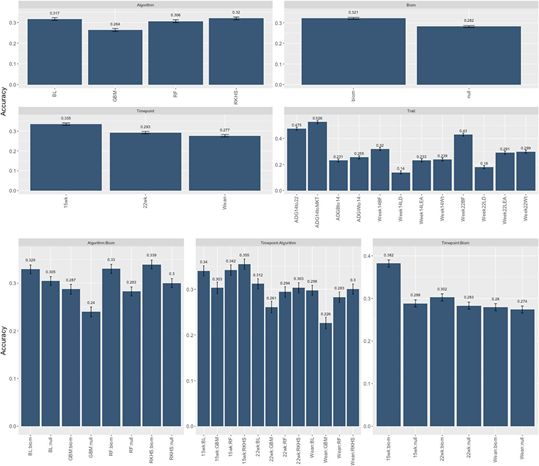
图6.最小二乘均值和SE的主要作用和相互作用,用于实验设计的后分析。时间点= 3个级别(断奶,15周,22周),算法= 4个级别(贝叶斯套索,再现核希尔伯特空间,随机森林,梯度增强机)特性= 12个级别(“ ADGBto14”,“ ADGWto14”,“ ADG14to22” ,“ ADG14toMKT”,“ Week14Wt”,“ Week14BF”,“ Week14LD”,“ Week14LEA”,“ Week22Wt”,“ Week22BF”,“ Week22LD”,“ Week22LEA”),Biom = 2个级别(无效,微生物组)。 所有带有(:)的元素表示成对交互。
图7.总体实验设计。BL =贝叶斯套索,RF =随机森林,GBM=梯度增强,RKHS =再现内核希尔伯特空间。 ADG =平均每日收益。
讨论
一般来说,我们的交叉验证强调了良好的预测能力,但结果因所考虑的时间点和特征而有很大差异。从我们的研究中,采样时间可能是将微生物群信息集成到生长预测模型中的关键因素。我们的数据表明,在生长试验中期测量的样品将提供最多的信息量。相反,微生物组组成的早期测量可能无法提供足够的信息。这与最近的研究有些不同,这些研究发现,在早期阶段,不同的肠型与生长特征有关。根据我们的经验,正如Lu等人所强调的,在早期时间点将个体聚集可能是仔猪或多或少迅速适应断奶时通常发生的饮食变化的结果。我们认为应该对此进行进一步调查。在本文中,我们认为每个时间点的研究都是独立的。这是一种简化,我们允许构建一个简单的交叉验证实验来测试不同的变量。尽管如此,今后使用纵向模型将提供一种更有力的方法来研究微生物群组成变化的重要性,以及这些变化如何影响牲畜的生长效率。
到目前为止,在纵向数据预测的背景下开发的一些深度学习模型应该可以更好地理解微生物组组成变化与表型结果之间的复杂相互作用。尽管如此,还需要更多的个体以及更深入的采样,才能达到必要的数据粒度,使这些方法具有吸引力。在我们的研究中,生长性状和脂肪性状均具有良好的预测能力。此外,当前的研究是在单个杂交种群中进行的。为了有效利用猪的微生物群落变异性,应调查更多的种群/品种,因为猪体内OTU成分的变异性较大。在这项工作中,我们建立了一个框架,以后可以扩展到不仅包括微生物组信息,还包括宿主基因组数据,以更好地表征和管理环境,并解决宿主和外来变异之间的复杂关系。微生物组组成可以有效地用作生长和组成性状的预测指标,尤其是对于脂肪性状。包含OTU预测因子可潜在地用于促进个体的快速生长,同时限制脂肪的积累。早期的微生物组指标可能不能很好地预测生长,而OTU信息则可能在生命的后期阶段得到最好的收集。应该注意的是,在当前的论文中,我们已经将微生物成分作为一个整体预测因素,并且我们并未尝试确定一个重要的OTU子集来减少预测因素的空间。这种方法将产生更强大和可移植的结果,特别是用于选择目的。但是,补充材料中报告了与时间/特征的每种组合显著相关的各个OTU的更多信息(补充表1)。
方法
动物。本研究中使用的猪在由The Maschhoffs LLC(美国伊利诺伊州卡莱尔)经营的商业环境中生长。因此,数据收集不需要动物使用许可。当前研究的后代来自二十八头纯种杜洛克种公,来自正在选择瘦肉的杜洛克种群,与大白×长白或长白×大白母猪交配。所产生的后代在18.6天(±1.09)断奶,然后转移到育苗场。
在这里,个体被分为20只猪一组。每组都是同父异母兄弟的姐妹,性别相同,体重相似。我们对这一基本实验模块进行了六次重复实验,每组均来自28头母猪,每支由2个组组成(雌性一组,一组被阉割的雄性)。试验期从猪进入育苗场的那天开始。在育苗期,生长期和肥育期向个体喂食标准颗粒饲料。提供了饮食配方及其营养价值[参见附加文件1]。猪接受了标准的疫苗接种和药物常规治疗。当组中的所有猪的平均活重达到136 kg时,以组为基础的测试结束。他们的平均收割年龄为196.4天(±7.86)。我们在三个时间点将组在所有猪中收集了直肠拭子:断奶,断奶后15周(平均118.2±1.18天,以下简称“ wk15”)和断奶后22周(平均196.4天±7.86天,之后) “wk22”)。每只猪随机选择四头猪进行瘦肉胴体生长测定,并将其直肠拭子用于微生物组测序。
最后,在第15周和第22周断奶时的样本数量分别为1205、1295和1283。 在所有三个时间点收集了1039只动物的样本。 提供了有关样本在家族,时间点和性别上分布的更多详细信息。 在14周和22周,在动物身上记录下腰深、腰部面积以及背部脂肪厚度和重量,并记录为商品重。这些措施在下文中分别称为Week14LEA,Week14LD,Week14BF,Week14Wt和Week22LEA,Week22LD,Week22BF,Week22Wt。 同样,从出生到第14周(ADGB14),从断奶到第14周(ADGW14)从第14周到第22周(ADG1422)和从第14周到上市(ADG14MKT)的活重差异来衡量平均日增重。 表3报告了当前分析中使用的特征摘要。
表1. 5倍交叉验证的特征/模型/年龄类别的每种组合的均方误差平均值和标准差。 BL =贝叶斯套索,RF =随机森林,GBM =梯度提升机,RKHS =复制内核希尔伯特空间。 ADGBto14 =到第14周的平均日增重,ADGWto14 =到第14周的平均日增重,ADG14to22 =第22周到第22周的平均日增重,ADG14toMKT =进入市场的第14周的平均日增重,Week14Wt = 第14周的体重, Week14BF = 第14周的背脂, Week14LD = 第14周的腰部深度, Week14LEA = 第14周的腰部眼肌, Week22Wt = 第22周的体重, Week22BF = 第22周的背脂, Week22LD = 第22周的腰部深度, Week22LEA =第14周的腰部眼肌.
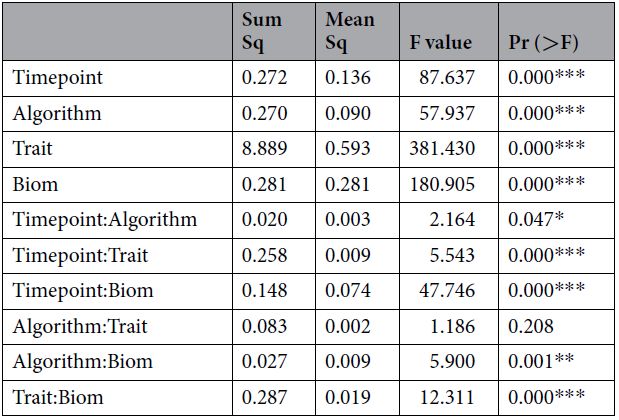
表2.实验设计的后分析ANOVA表。 时间点= 3个级别(断奶,15周,22周),算法= 4个级别(贝叶斯套索,再现内核希尔伯特空间,随机森林,梯度增强)特性= 12个级别(“ ADGBto14”,“ ADGWto14”,“ ADG14to22”, “ ADG14toMKT”,“ Week14Wt”,“ Week14BF”,“ Week14LD”,“ Week14LEA”,“ Week22Wt”,“ Week22BF”,“ Week22LD”,“ Week22LEA”),生物素= 2级(无效,微生物组)。 所有带有(:)的行均表示成对交互。
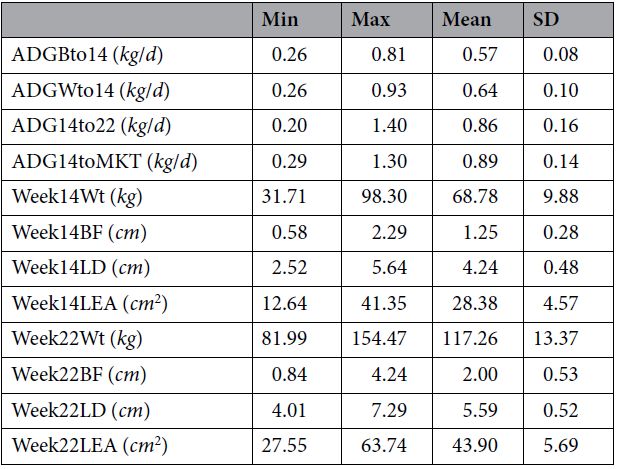
表3.研究中使用的表型摘要。 ADGBto14 =到第14周断奶的平均日增重,ADGWto14 =到第14周断奶的平均日增重,ADG14to22 =第14周的平均日增重,第22周,ADG14toMKT =进入市场14的平均日增重,Week14Wt = 第14周的体重, Week14BF =第14周的背脂, Week14LD =第14周的腰部深度, Week14LEA =第14周的腰部面积 , Week22Wt=第22周的体重, Week22BF =第12周的背脂, Week22LD= 第22周的腰部深度, Week22LEA =第22周的腰部面积.
DNA提取和纯化。通过苯酚:氯纤维的机械破坏从每个直肠拭子中提取总DNA(gDNA)。简而言之,将650 pL提取缓冲液(200 mM Tris; 200 mM NaCl; 20 mM EDTA,pH 8.0)添加到每支拭子中,这些拭子存储在2 mL自立式螺旋盖管(Axygen,CA,美国)中。使用Mini-BeadBeater-96(MBB-96; BioSpec,OK,美国)摇动试管20秒钟,以从拭子头中取出样品材料。短暂离心(10 s;500×g)以拉下所有脱落的物质后,使用无菌镊子将每个拭子头从其试管中取出。将样品在-80°C下冷冻成固体,然后将大约250 pL的0.1 mm氧化锆/二氧化硅珠(BioSpec)和3.97 mm不锈钢球添加到样品中(仍保持冷冻以避免飞溅)。使样品短暂融化,然后加入210 pL 20%SDS和500 pL苯酚:氯仿:IAA(25:24:1,pH 8.0)。在MBB-96上进行珠击(4分钟;室温),将样品离心(3220×g; 4分钟),然后将250 pL水相转移至新试管中。然后使用QIAquick 96 PCR纯化试剂盒(Qiagen,MD,USA)进一步纯化100 pL粗DNA。按照制造商的说明进行纯化,但需进行以下较小改动:(i)将乙酸钠(3 M,pH 5.5)添加到缓冲液PM中,终浓度为185 mM,以确保基因组DNA与硅胶膜的最佳结合; (ii)将粗DNA与4体积的Buffer PM(而不是3体积)混合; (iii)用100 pL Buffer EB(而不是80 pL)洗脱DNA。
Illumina文库的制备和测序。 使用Faith等描述的策略,对16S rRNA基因的V4区(515-806)进行了分阶段的双向扩增,以生成用于Illumina测序的索引文库。 在以等摩尔比合并之前,使用Qubit dsDNA测定试剂盒(Thermo Fisher Scientific Inc.,MA,USA)对扩增子文库进行定量。 根据制造商的说明,使用Agencourt AMPure XP磁珠(Beckman Coulter)纯化这些最终合并液。纯化池辅以 5-10% PhiX 控制 DNA,并使用 600v3 试剂盒在 Illumina MiSeq 机器上测序成成对端 2x250 + 13 bp指数反应。未解的 FASTQ 文件由 MiSeq 报告器生成。所有测序均在圣路易斯华盛顿大学基因组科学与系统生物学中心的DNA测序创新实验室进行。
16S rRNA基因测序和数据质量控制。首先使用FLASh v1.2.11将成对的V4 16S rRNA基因序列对合并为单个序列,要求重叠至少100个且不超过250个碱基对,以提供可靠的重叠。然后使用PRINSEQ v0.20.449滤出平均质量得分低于Q35的序列。序列以正向定向,并且任何引物序列都经过匹配和修剪;在引物匹配期间,最多允许1个错配。序列随后使用 QIIME v1.950 进行多路复用。然后使用具有以下设置的QIIME将具有> 97%核苷酸序列同一性的序列聚类为可操作的分类单位(以下称“ OTU”):max_accepts = 50,max_rejects = 8,percent_subsample = 0.1和--suppress_step4。 GreenGenes的修改版本(Greengenes数据库联盟51-53)被用作参考数据库。然后,将具有10%读取但未命中参考数据库的输入序列与UCLUST54从头进行聚类,以生成新的参考OTU,其余90%的读取将分配给这些OTU。每个簇中最丰富的序列用作OTU的代表性序列。然后通过保留最少的总观察计数1200以保留OTU来过滤掉稀疏的OTU,并且每个样品的OTU表被稀疏到10,000个计数。在分在15周和第22周,平均良好值的覆盖率估计值分别为0.99±0.002、0.98~0.002和0.98±0.002。最后,核糖体数据库计划(RDP)分类器(v2.4)按照Ridaura及其同事所述的方式进行了重新训练,其中使用0.8阈值将分类法分配给代表性序列。经过数据处理和质量控制后,可以使用1755个OTU进行进一步分析。
统计分析。 培训和测试集。 使用分层的五重交叉验证方案将数据递归随机地分为训练(约占观察值的70%)和预测(约占观察值的30%),并保持试验中28个父本的均等代表。 总体实验设计的图形表示如图2所示。
模型。 所有模型均在回归框架中用于我们分析中的模型。 为了进行研究,将方法,特征和时间的每种组合作为一个单独的分析,并将每个模型的预测准确性作为测试集中预测表型与测量表型之间的平均皮尔逊相关性来获得,类似于在全基因组预测研究中提出的。此外,还获得了平方误差及其标准偏差。
贝叶斯套索。 对于每种折叠/特征/时间点组合,都安装了两个模型:
空模型(null):
y = µ+ Xb + e
其中:y是上一节提到的特征之一,μ是总体平均值,b是固定效应的向量,包括:性别(2个层次),重复(6个层次),父亲(28个层次)以及协变量断奶时的重量e是假定N(0,)的随机残差的向量,而X是将观察值与固定效应相关的入射矩阵。
包含微生物组(biom)的模型:y = µ + Xb +Wo + e
其中:o 是 OTUs 效果(1755级)的矢量,W 是居中和缩放的 OTUs 计数的矩阵,其余与前一个模型中相同。
我们拟合了R软件包BGLR实施的BL回归模型。 OTU计数通过使用双指数先验分布拟合到模型。BGLR将双指数密度建模为按比例缩放的正常密度。在层次结构的第一级中,标记效果被分配了具有零均值和特定于OTU的方差参数T2xaf的独立法线密度。残余方差被指定为比例倒数卡方先验密度。 BGLR提供了一种通过R2标志选择先验形状的便捷方法。 R2可以大致解释为模型中包含的效应所解释的预期方差比例。对于残余效应,默认自由度为5,R2为0.60。然后获得先验尺度参数,即Sp = Var(y)(1-R2)(dfp + 2),其中Sp和dfp分别为尺度和自由度。 OTU的特定比例参数T2分配了IID指数密度,速率参数为2/2。在这种情况下,超参数入是固定的,并且其值通过对整个数据集/特征组合进行网格搜索来分配(结果未显示)。
随机森林。这里采用的空模型的一般形式是(遵循Gonzalez-Recio和Forni):
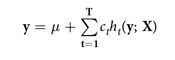
而生物模型是:
每个树ht(y;X) 或 ht(y;t G (1, T) 的 X + W)由原始数据的随机样本构造,在每个节点上随机选择要素子集以创建拆分规则。每棵树都尽可能地生长,直到所有终端节点都最大地均匀。参数[if !msEquation] [endif]是平均树的收缩系数。RF 中的分割质量可以通过不同的标准进行测量。对于当前分析,采用均方误差(MSE)。这项工作中RF模型的剩余参数设置如下:(i) 树的数量设置为等于1500;(ii) 查找最佳拆分时要考虑的要素数等于原始要素数的根数。R的 bigrf 封装用于将 RF 型号适合数据。
渐变提升。此处采用的空模型的一般形式是(再次跟随Gonzalez-Recio 和 Forni):
而生物模型是:
在这种情况下,将t G(1,M)的每个预测变量hm(y; X)或hm(y; X + W)依次应用于先前的组所形成的委员会的残差,装袋步骤保持类似到之前描述的内容。 R的gbm package用于将GBM模型拟合到数据。 使用高斯损失函数。 GBM模型中的其他参数设置如下:i)树的数量设置为1500; ii)交互深度设置为3; iii)将收缩参数v设定为0.01。
复制内核希尔伯特空间。安装了两种 RKHS 型号:
空模型 (空):y = µ+ Xb + e
和 (生物)形式模型:y = µ + Xb + Zu+ e
其中Z 是一个入射对角矩阵的顺序 (1039 × 1039) 和 u是假定N(0,Mσu2))的猪效应的随机矢量。 M是基于微生物群落成分的内核矩阵,其计算如下:在OTU级别使用微生物群来计算样本对之间的Jensen-Shannon距离,
其中 D(a、 b) 是样本a和 b之间的距离; n 是OTUS 数量(n = 1755); ai 和 bi分别是样本a 和 b中的 OTUi 计数; mi=(ai=bi)/2.生成的方形矩阵(下称"JSD")对角线上为零,对角线值介于 0 和 1 之间。M矩阵作为1-JSD得到。 RKHS回归模型在贝叶斯设置内使用 R 包 BGLR 实现。之前为[u2],如果选择在上一节中突出显示。两个参数的 R2 值分别设置为 0.3 和 0.6。
后分析。 为了提供对设计中所有因素的全面评估,我们使用标准的线性混合模型(LMM)对实验进行了后期分析。 复制/特征/方法的所有组合都集中在一个数据集中。 然后安装了以下LMM
yijklm是每个重复/特征/方法组合的准确性; Ti是微生物组时间点测量的固定效果(3个级别:断奶,15周,22周); Aj是所使用算法的固定效果(4个级别:BL,RKHS,RF,GBM); Trk是特质的固定效应(12个级别:ADGBto14,ADGWto14,ADG14to22,ADG14toMKT,Week14Wt,Week14BF,Week14LD,Week14LEA,Week22Wt,Week22BF,Week22LD,Week22LEA); B1是微生物组包含物的固定作用(2个级别:无效,生物组); TAij TTrik TBil ATrjk和ABjl是主要效应的成对相互作用。 TATrBijkl是假设N(0,
)的T,A,Tr和B的随机相互作用效应; eijklm是假设N(0,σ2)的随机残余效应。 LMM模型装有R软件包lme4。 用R包lmerTest获得III型ANOVA表,最小二乘均值和对比度。