轨迹分析_Scanpy进行单细胞分析及发育轨迹推断
分享是一种态度

最近看文献,发现越来越多的单细胞测序使用scanpy进行轨迹推断,可能因为scanpy可以在整体umap或者Tsne基础上绘制细胞发育路径,图片也更加美观,但是Scanpy是基于python开发的,下面整理下Scanpy官网给出的流程,按照官网流程跑一遍PBMC的数据。
推荐大家使用anaconda中的jupyter进行相关分析,非常便于数据的复现以及随时矫正~
在jupyter使用pip install scanpy 完成scanpy安装。
Scanpy对于10X cellranger的pipline数据可以直接读取,打开python后:
import numpy as np
import pandas as pd
import scanpy as sc
sc.settings.verbosity = 3
sc.logging.print_versions()
results_file = 'pbmc3k.h5ad' #相关结果都保存在这个文件下
adata=sc.read_10x_mtx('/project/Scseq/filtered_gene_bc_matrices/hg19', var_names='gene_symbols', cache=True) #读取单细胞测序文件
sc.pl.highest_expr_genes(adata, n_top=20,save="counts.pdf" )
#可以在命令里加入save="***.pdf" 来保存相关图片
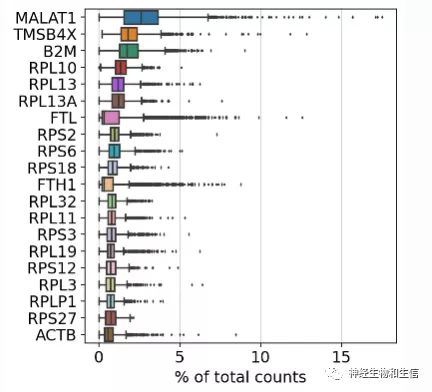
#显示在所以细胞中那些基因的count数最多
adata.var_names_make_unique()
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
#对于细胞内基因数少于200的或者低于3个细胞表达的基因去除
adata.var['mt'] = adata.var_names.str.startswith('MT-') # annotate the group of mitochondrial genes as 'mt'
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'], jitter=0.4, multi_panel=True)
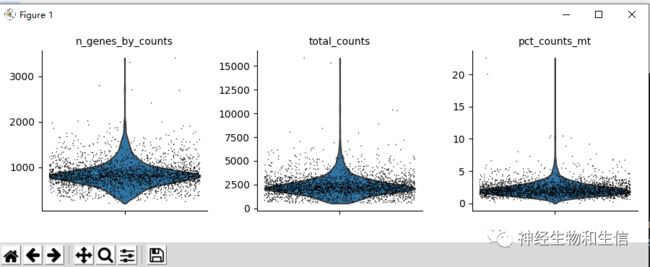
- 每个细胞中表达的基因数
- 每个细胞中的counts数
- 线粒体基因百分比
下一步我们删除表达太多线粒体基因或counts过多的细胞
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')
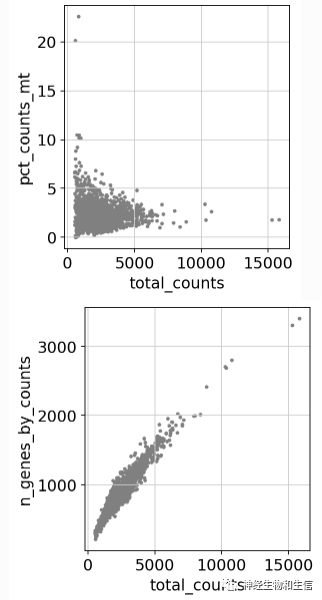
adata = adata[adata.obs.n_genes_by_counts adata = adata[adata.obs.pct_counts_mt 对数据进行进一步过滤
当然过滤的时候要考虑实际情况,比如如果你做的实验细胞本身线粒体含量就比较高,那么就没必要过滤>5%线粒体基因的细胞了。
sc.pp.normalize_total(adata, target_sum=1e4)
#标准化数据
sc.pp.log1p(adata)
#鉴定高变基因
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
## 提取高度variable的基因
sc.pl.highly_variable_genes(adata)
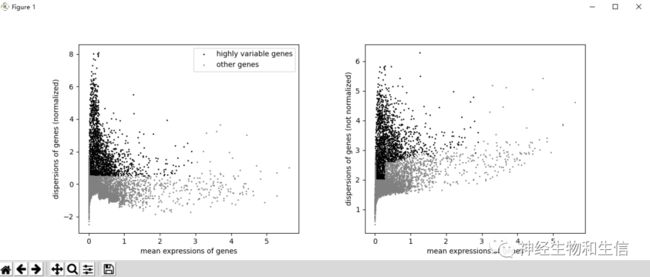
adata.raw = adata
adata = adata[:, adata.var.highly_variable]
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
sc.pp.scale(adata, max_value=10)
进一步进行数据过滤
排除掉线粒体基因,细胞总counts数对下游PCA的影响
sc.tl.pca(adata, svd_solver='arpack')
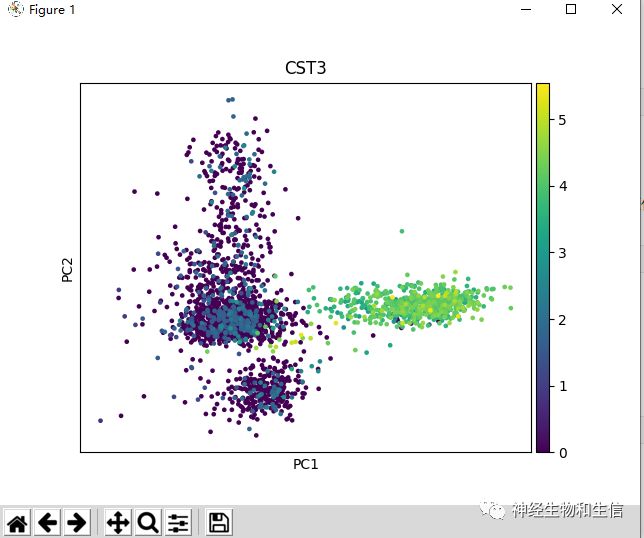
sc.pl.pca(adata, color='CST3')
#在高可变基因进行PCA
sc.pl.pca_variance_ratio(adata, log=True)
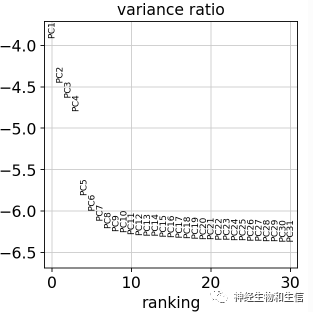
减少干扰因素
确定有多少个PCA会对下游产生影响,可以看到PCA到15以后就趋于水平,那么在下游进行TSNE或UMAP细胞分群时候,我们就可以只选取前15个PCA结果,这样减少干扰因素。
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
#Computing the neighborhood graph
sc.tl.umap(adata)
## 计算UMAP,细胞分群
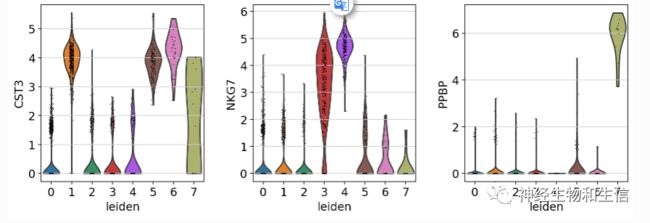
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'])

sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)
使用标准化后的数据进行绘图
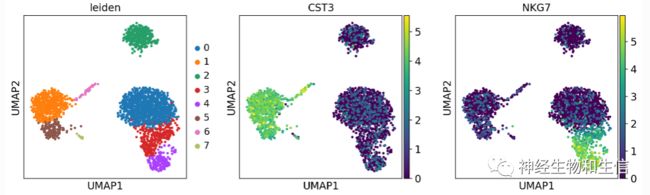
sc.tl.leiden(adata)
使用leiden进行聚类
别忘记提前在python上安装相关算法
sc.pl.umap(adata, color=['leiden', 'CST3', 'NKG7'])
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
寻找marker基因
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
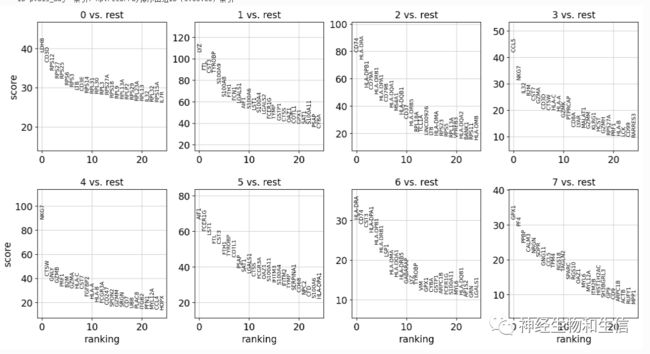
非常直观的给出了每一个cluster里的marker基因排名
sc.settings.verbosity = 2 # reduce the verbosity
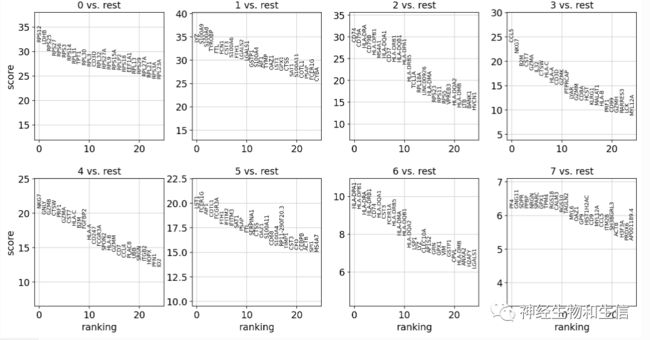
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
# Wilcoxon秩和
sc.tl.rank_genes_groups(adata, 'leiden', method='logreg')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
marker_genes = ['IL7R', 'CD79A', 'MS4A1', 'CD8A', 'CD8B', 'LYZ', 'CD14',
'LGALS3', 'S100A8', 'GNLY', 'NKG7', 'KLRB1',
'FCGR3A', 'MS4A7', 'FCER1A', 'CST3', 'PPBP']
定义marker基因列表
pd.DataFrame(adata.uns['rank_genes_groups']['names']).head(5)
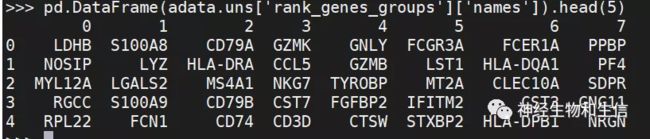
在数据框中显示每个cluster 0、1,…,7的前10个排名最高的基因。
sc.tl.rank_genes_groups(adata, 'leiden', groups=['0'], reference='1', method='wilcoxon')
sc.pl.rank_genes_groups(adata, groups=['0'], n_genes=20)
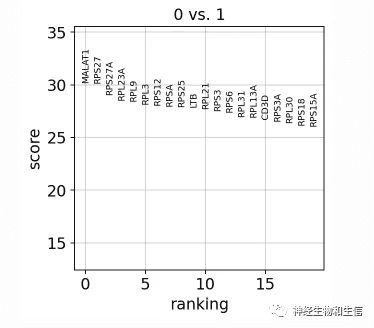
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
某两个组比较
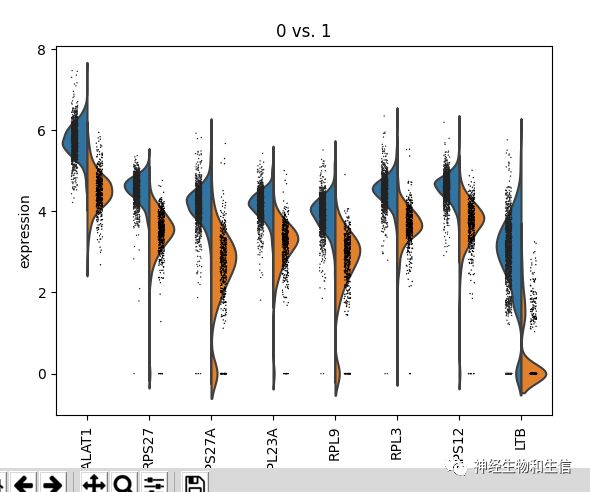
sc.pl.violin(adata, ['CST3', 'NKG7', 'PPBP'], groupby='leiden')
跨组比较某个基因
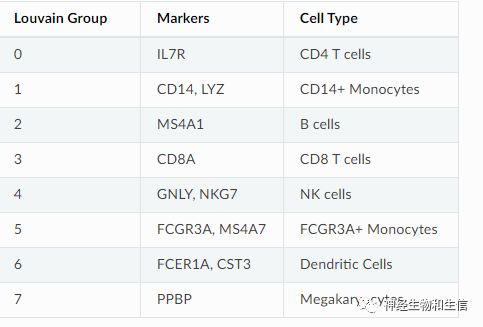
new_cluster_names = [
'CD4 T', 'CD14 Monocytes',
'B', 'CD8 T',
'NK', 'FCGR3A Monocytes',
'Dendritic', 'Megakaryocytes']
adata.rename_categories('leiden', new_cluster_names)进一步对分群进行标记
sc.pl.umap(adata, color='leiden', legend_loc='on data', title='', frameon=False, save='.pdf')

sc.pl.dotplot(adata, marker_genes, groupby='leiden');
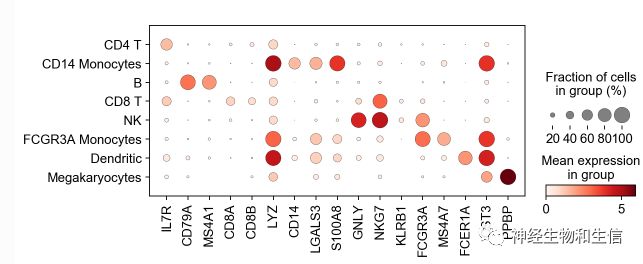
可视化标记基因
sc.pl.stacked_violin(adata, marker_genes, groupby='leiden', rotation=90);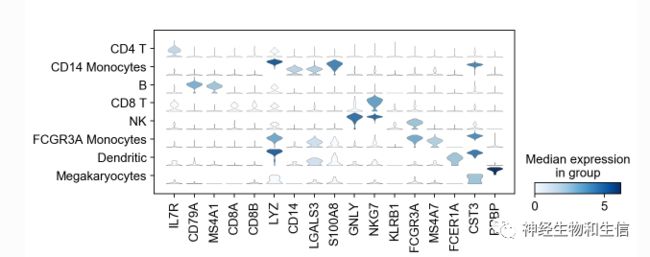
小提琴图显示
到目前为止已经完成了上游分析,保存方式分为三种。
1,保存所有的分析及内容
adata.write(results_file, compression='gzip') # `compression='gzip'` saves disk space, but slows down writing and subsequent reading
2,保留原始数据,删除 scaled and corrected data matrix。
adata.raw.to_adata().write('./write/pbmc3k_withoutX.h5ad')
3.导出CSV格式数据
# Export single fields of the annotation of observations
# adata.obs[['n_counts', 'louvain_groups']].to_csv(
# './write/pbmc3k_corrected_louvain_groups.csv')
# Export single columns of the multidimensional annotation
# adata.obsm.to_df()[['X_pca1', 'X_pca2']].to_csv(
# './write/pbmc3k_corrected_X_pca.csv')
# Or export everything except the data using `.write_csvs`.
# Set `skip_data=False` if you also want to export the data.
# adata.write_csvs(results_file[:-5], )
下一步就是完成逆时序列分析
首先设置相关参数并导入之前分析的数据
import numpy as np
import pandas as pd
import matplotlib.pyplot as pl
from matplotlib import rcParams
import scanpy as sc
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_versions()
results_file = './paul15.h5ad'
sc.settings.set_figure_params(dpi=80, frameon=False, figsize=(3, 3), facecolor='white') # low dpi (dots per inch) yields small inline figuresadata = sc.read( 'C:\\Users\\biostack\\pbmc3k_withoutX.h5ad')
adata.X = adata.X.astype( 'float64') #float32是一个32位数字 – float64使用64位.这意味着float64占用了两倍的内存 – 在某些机器架构中对它们进行操作可能会慢得多,但是,float64可以比32位浮点数更准确地表示数字,它们还允许存储更大的数字。
对数据进行预处理
sc.pp.recipe_zheng17(adata)
sc.tl.pca(adata, svd_solver='arpack')
sc.pp.neighbors(adata, n_neighbors=4, n_pcs=20)
sc.tl.draw_graph(adata)
初步的绘图
sc.pl.draw_graph(adata, color='leiden', legend_loc='on data')
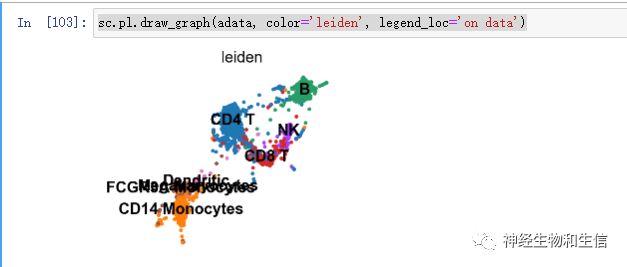
可以看到目前相对图形比较混乱,下一步有可供选择的降噪处理
sc.tl.diffmap(adata)
sc.pp.neighbors(adata, n_neighbors=10, use_rep='X_diffmap')
sc.tl.draw_graph(adata)
sc.pl.draw_graph(adata, color='paul15_clusters', legend_loc='on data')
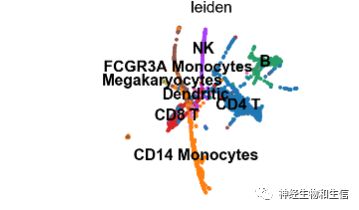
可以看到相比较于上一副图,有很多分化的分支已经被绘制出来。
同时scanpy可以进行PAGA轨迹推断
sc.tl.louvain(adata, resolution=1.0)
sc.tl.paga(adata, groups='louvain')
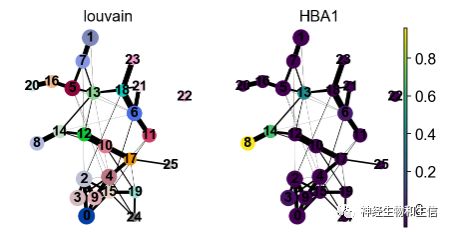
sc.pl.paga(adata, color=['louvain' , "HBA1"])
非常好用的一个功能,可视化某个基因的分布,这里可以选择一些跟分化相关的基因,这里为了区别不同的cluset,选择了血红蛋白相关基因HBA1
sc.pl.paga(adata, color=['louvain' , 'LYZ'])
选择另外一个单核细胞的marker-LYZ可视化,可以看到随着分化LYZ的表达量越来越高5-7-1.
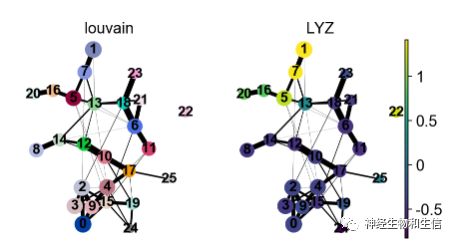
使用带有注释的分群
adata.obs['louvain'].cat.categories
adata.obs['louvain_anno'] = adata.obs['louvain']
sc.tl.paga(adata, groups='louvain_anno')
sc.pl.paga(adata, threshold=0.03, show=False)
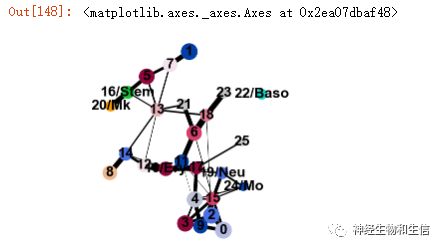
进一步在单细胞水平可视化相关基因分布
sc.tl.draw_graph(adata, init_pos='paga')
sc.pl.draw_graph(adata, color=['louvain_anno', 'CD14', 'CD8A', 'LYZ'], legend_loc='on data')
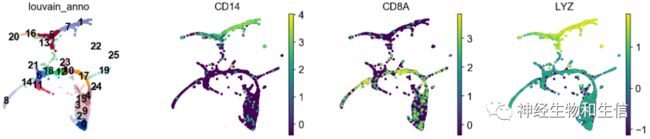
到目前已经完成了初步的分析,由于scanpy是建立在python基础上,事实上在使用中速度比R要快特别多~
 往期回顾
往期回顾
DESeq2差异表达分析(二)
cellphonedb 及其可视化
如果你对单细胞转录组研究感兴趣,但又不知道如何入门,也许你可以关注一下下面的课程
单细胞初级8讲和高级分析8讲
老板,请为我配备一个懂生信的师兄
你以为GEO只是挖挖就完了吗

![]()
看完记得顺手点个“在看”哦!
 生物 | 单细胞 | 转录组丨资料 每天都精彩
生物 | 单细胞 | 转录组丨资料 每天都精彩
长按扫码可关注