最近TCGA更新了,下载研究一下,我们从TCGA下载STAD的数据,选择其中的一个打开,发现了一个好消息那就是矩阵的整合难度降低了,而且提供TPM以及FPKM 还有校正的count 以及gene_name
在我的主页更新了TCGAbiolinks的方法,更为方便和快捷。同时我也提供了临床数据的处理方式
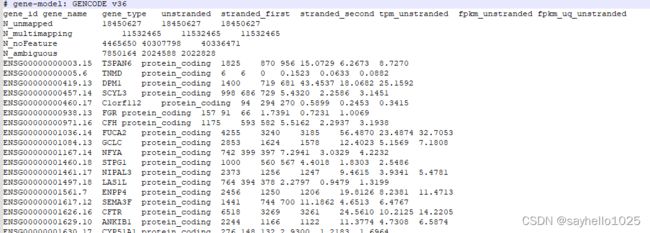
其实整理起来比较简单,这里我没有使用python去写脚本,使用R硬刚,说实话头有点铁。
首先整理好你要的数据,下载步骤跟以前一样,当然不懂就去B站看新版TCGA介绍
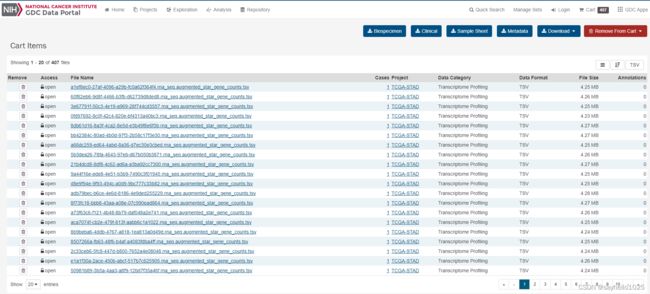
首先下载metadata
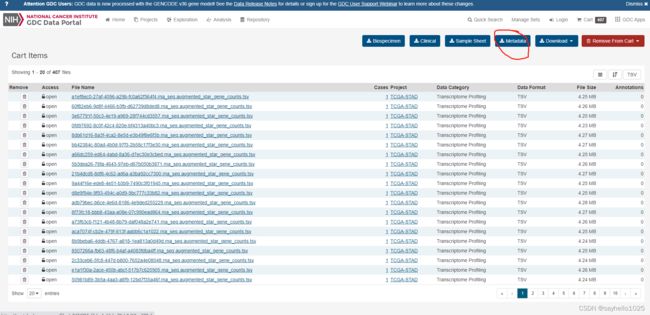
接着从download 中下载cart
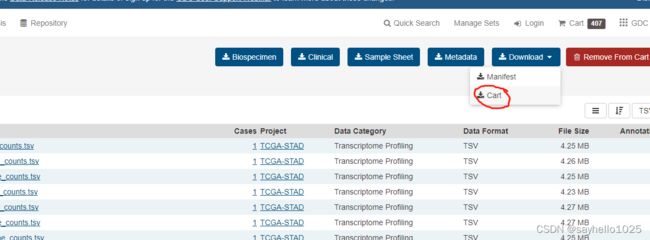
下载结束 之后解压随便打开一个文件,我们的目的是整合tsv文件到一个文件夹

技巧教授如何整合呢,windows下最简单的方式,右上角搜索.tsv结尾的文件
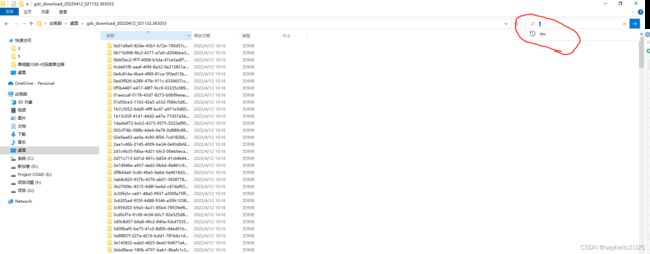
最后你就可以得到这个列表,然后复制到一个文件夹里面,最后有个MANIFEST.txt不要复制进去
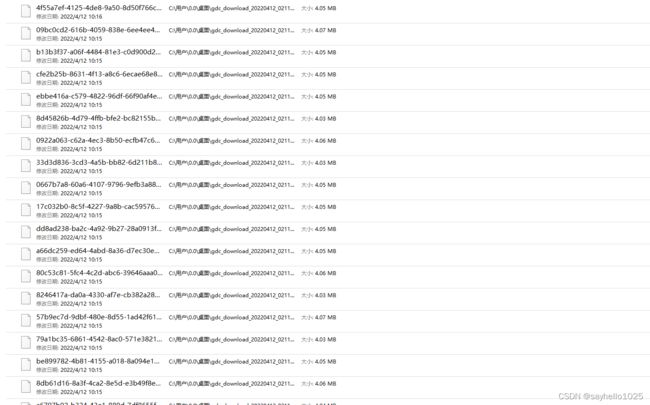
最后整理成下面的样式,all文件夹是所有的tsv文件,metadta上面介绍已经下载好了放进去
整理metadata文件
rm(list = ls())
options(stringsAsFactors = F)
library("rjson")
result <- fromJSON(file = "./metadata.cart.2022-04-12.json")
metadata <- data.frame(t(sapply(result,function(x){
id <- x$associated_entities[[1]]$entity_submitter_id
file_name <- x$file_name
all <- cbind(id,file_name)
})))
metadata[,2]
rownames(metadata) <- metadata[,2]
得到样本文件对应的TCGA的ID号
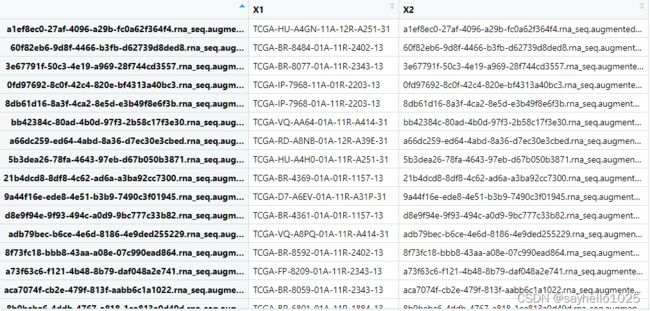
生成矩阵
dir <- './all/'
samples=list.files(dir)
sampledir <- paste0(dir,samples)
mat <- do.call(cbind,lapply(sampledir, function(x){
rt <- data.table::fread(x,data.table = F)
rownames(rt) <- rt[,1]
rt <- rt[,7]###后续方便不用再转换直接拿TPM
}))
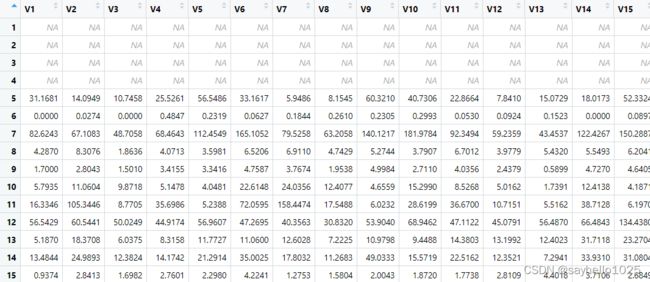
这里需要读取一个单个tsv文件进行查看
可以看到我们上面代码中选择rt[ ,7] 是选择的tpm ,需要的话可以你可以选择count和FPKM
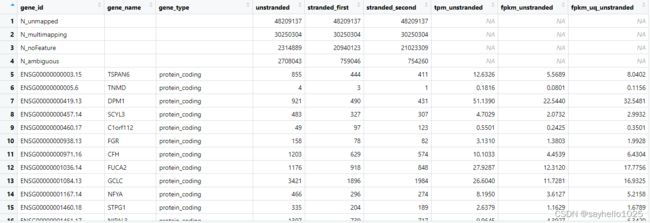
矩阵列明和行名的替换
###随便打开一个tsv文件即可
rt <- data.table::fread('./all/5c84144f-68b0-4f96-8b53-5a43a6f83847.rna_seq.augmented_star_gene_counts.tsv',data.table = F)###随便打开一个tsv文件即可
colnames(mat)=sapply(strsplit(sampledir,'/'),'[',3)###列名
rownames(mat) <- rt$gene_id ##行名
mat1 <- t(mat)
same <- intersect(row.names(metadata),row.names(mat1))
data <- cbind(metadata[same,],mat1[same,])
rownames(data) <- data[,1]
tcga_stad <- t(data)
tcga_stad <-tcga_stad[-c(1:6),]
rownames(rt) <- rt[,1]
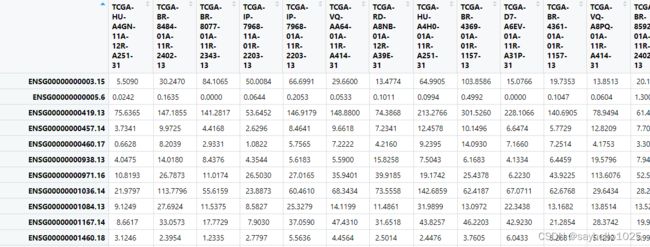
最后那必须是如何ID的转换 ,根据自带的信息我们进行转换,这次的更新就是ID给了对照基因名,真好,还有类型,妈的TCGA良心一次更新
same2 <- intersect(row.names(rt),row.names(tcga_stad))
tcga <- cbind(rt[same2,],tcga_stad[same2,])
tcga <- tcga[-c(1,4:9)]
到此你们自己保存文件把,然后正常的基因去重问题,这个应该搜索一下, 也可以再我的博客里面看看
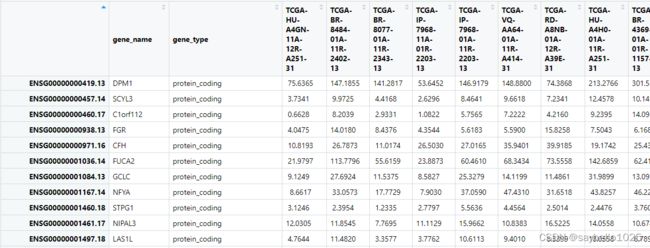
##要把上面的gene_type删除
rt = tcga[,-2]
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)
data=data[rowMeans(data)>0,]
到此你们应该得到都会处理的矩阵了把