单细胞多模态GAN揭示三阴性乳腺癌的空间模式

目录
- 摘要
- 引言
- 结果
-
- scMMGAN整体架构
- 空间、scRNA-seq和蛋白质组学数据之间的映射
- RNA-seq和ATAC-seq之间的映射
- 三阴性乳腺癌数据的整合
- 局限
摘要
生物实验数据越来越多地沿着多个不同的模态生成,每年都在开发专门用于特定测量的新的和更复杂的技术。使用多种专用数据收集工具测量单个对象或系统,自然会产生将这些单独工具的结果整合成单个统一视图的想法。论文介绍了为此目的设计的计算技术。使用单细胞多模态GAN(scMMGAN,single-cell multi modal GAN),沿许多不同的组学方向进行计算,并将每个方向的信息合成为对所研究系统的更大视图。
测量生物系统技术的进展目前处于研究的前沿。沿着越来越多的组学维度收集数据的能力已经逐渐实现,我们想将所有这些信息分析在一起,而不是将每一项技术孤立地放在单独的分析管道中。为了推进这一目标,我们引入了一个称为单细胞多模态GAN(scMMGAN)的框架,该框架将来自多种模态的数据整合到统一表示中,以便进行下游分析。该框架的关键改进是额外的扩散几何损失(diffusion geometry loss),新的核约束了过度参数化的GAN。我们证明了scMMGAN在多种数据模态上产生比替代方法更有意义的比对能力,其输出可用于从真实生物实验数据得出结论。
引言
整合从不同来源收集的数据是计算基因组学的一个关键挑战。目前有几种单细胞测序技术,包括RNA测序(RNA-seq)、使用测序技术(ATAC-seq)测定转座酶可接近的染色质、Hi-C、ChIP-seq(染色质免疫沉淀测序)和CITE-seq(通过测序对转录组和表位的细胞索引)以及蛋白质组学技术,例如飞行时间流式细胞术 (CyTOF)、imaging CyTOF 和提供互补细胞信息的多路离子束成像。在这些模态中,只有一小部分可支持同时测量——并且通常具有质量退化现象,例如基因尺寸减小、通量降低和噪声增加。其余的测量必须在同一群落的不同细胞样本上进行。这是我们在本文中解决的关键问题:预测缺失或非同时测量的模态,以便生成更完整的数据集。因此,给定在不同细胞(来自同一群落)上分别测量的单细胞RNA-seq(scRNA-seq)、scATAC序列和空间转录组学等模态,我们的单细胞多模态生成对抗网络(scMMGAN)为下游分析生成了一套完整的同时测量的数据(配对数据)。
与单独分析某个模态相比,在计算上对齐各模态单独测量的数据具有许多优点。组合模态使我们能够充分利用每种模态的优点。例如,将具有较高信噪比(如蛋白质组学细胞因子测量)的模态与具有较低信噪比的模态(如scRNA-seq)相结合,我们有机会在噪声较大的数据集中更精细地解析细胞群。更令人信服的是,组合模态允许我们测量仅在一个域中可用的变量与仅在另一个域可用的变量组合,从而模拟联合测量技术。
我们的方法基于循环一致生成对抗网络(CycleGANs)的框架。在基于GAN的域适应框架中,生成器网络被训练为将一种模态的数据点映射到另一种模态中的数据点。在训练期间,使用鉴别器来检验从第二模态的高维分布中采样生成的点。在CycleGANs中,有两个背靠背的生成器,一个从第一模态到第二模态,另一个从第二模态到第一模态。重建误差要求两个背靠背域适应的结果接近原始分布。
虽然CycleGAN框架可以在每个模态中成功生成点,但它们生成的映射没有受到足够的限制。例如,在最初的CycleGAN论文中,斑马的图像被映射到马的图像,但没有任何东西可以确保背景不会被改变。虽然这可能不会对自然图像应用造成不利影响,但对于scRNA-seq测量必须与scATAC-seq测量相对应并加以证实的科学应用来说,这可能是站不住脚的。注意到这一关键弱点,我们指定了一个更强大、更普遍适用的对应损失:几何保持损失(geometry-preserving loss)。这种损失使扩散几何(diffusion geometry)在整个映射过程中得以保留,该几何体是用一个新的核来执行的,该核旨在传递比高斯核更好的梯度。我们注意到,即使在没有重叠测量的情况下,也可以利用这种损失。
我们演示了在各种数据类型上使用scMMGAN调整数据模态的能力。我们首先在同时测量可用的数据集上验证它,并将其用作评估的Ground Truth。然后,我们使用scMMGAN对一个新的三阴性乳腺癌数据集进行了彻底的研究,其中我们将来自乳腺癌培养物HCC38的细胞移植到小鼠中,并允许其从原发肿瘤转移到继发肿瘤位置。我们表明,scMMGAN可以推断细胞结构的空间位置。
结果
scMMGAN整体架构
scMMGAN框架如图1A所示。每一对数据域或模态都有一对生成器网络,它们在它们之间的任意方向上映射,形成多样化的多模态映射。对于从域 i i i到域 j j j的生成器映射,我们标记为 G i j G_{ij} Gij,对应在域 j j j上的判别器记为 D j D_{j} Dj,用于区分来自域 j j j的真实数据 x j x_{j} xj和生成器产生的数据 G i j ( x i ) G_{ij}(x_{i}) Gij(xi)。而生成器需要生成逼真的数据蒙蔽判别器。它们交替优化下面目标: m i n G i j m a x D j E x j ∼ P ( x j ) l o g ( D j ( x j ) ) + E x i ∼ P ( x i ) l o g ( 1 − D j ( G i j ( x i ) ) ) min_{G_{ij}}max_{D_{j}}E_{x_{j}\sim P(x_{j})}log(D_{j}(x_{j}))+E_{x_{i}\sim P(x_{i})}log(1-D_{j}(G_{ij}(x_{i}))) minGijmaxDjExj∼P(xj)log(Dj(xj))+Exi∼P(xi)log(1−Dj(Gij(xi)))判别器引导生成器将其输入模态转换为输出模态,上述GAN的损失记为 L G A N L_{GAN} LGAN,另外还有两个其他term,确保学习的映射信息丰富且有意义。如图1B所示。重建损失 L r L_{r} Lr是原始数据 x i x_{i} xi和域 i i i和 j j j之间的两个成对生成器的组成之间的均方误差(MSE): L r = ∣ ∣ x i − G j i ( G i j ( x i ) ) ∣ ∣ 2 L_{r}=||x_{i}-G_{ji}(G_{ij}(x_{i}))||^{2} Lr=∣∣xi−Gji(Gij(xi))∣∣2对应损失(correspondence loss) L c L_{c} Lc对域 i i i和域 j j j中的single point的representation施加约束,而重建损失对同一域中的点施加约束: L c = c o r r e s p o n d e n c e ( x i , G i j ( x i ) ) L_{c}=correspondence(x_{i},G_{ij}(x_{i})) Lc=correspondence(xi,Gij(xi))对应损失 L c L_{c} Lc的动机来自于这样一个事实,即先前使用循环一致性进行GAN域映射的模型包括两个限制:
- 生成器能够在点移动到另一个域并返回原域的重建点;
- 判别器不能区分真实点和映射点。
生成器可以通过许多不同的方式实现这些目标,包括学习任意复杂的映射:只要它们在分布级别对齐两个数据流形。可以匹配目标分布的成对函数族很多,并且在现有框架中,训练产生的由随机权重初始化和梯度下降中的神秘偏差的变化很难控制。
然而,简单匹配概率分布可能会导致非相干细胞状态(图1D)。我们带来的一个关键见解是,分布必须仅在对应约束内匹配。这些对应关系本质上是基础系统中的不变性(invariances),反映在每个模态中。在之前的工作中,我们为每个数据集使用了定制的对应损失。然而,这里我们注意到,当匹配单细胞数据时,我们可以使用几乎通用的约束,即流形几何保持。
虽然我们的模型将来自数据几何的信号合并到更大的框架中,但数据几何过于僵硬,无法单独用于指导对齐。它严重受域数据空间属性的影响,因此,当两个域非常不同时,它不允许在改变分布形状方面具有足够的灵活性。仅使用数据几何的方法难以对齐显著不同的域。理想的映射将具有匹配概率分布的映射的灵活性(就像 GAN 所做的那样),但在这样做的同时尽可能地保留数据几何形状。通过结合现有的GAN的基本loss和数据几何loss,网络可以平衡这些目标之间的权衡。
因此,我们引入了一种correspondence loss,通过学习映射来保持数据几何结构,从而确保映射具有point-wise的分布对齐。为此,我们使用原始数据的diffusion map表示。
这里,我们简要概述diffusion map。扩散映射(diffusion map)是流形学习中常用的基于核的方法,用于生成低维嵌入,以保留数据中的固有结构(基于样本距离进行计算)。扩散算子的特征向量形成embedding,其中欧几里德距离对应于原始流形上的扩散距离,或通过随机游走从一点到另一点的概率。因为由扩散特征向量表示的这些新坐标抽象了许多数据域特定属性,提供了一种确保在映射中保留底层数据几何结构的方法。

- 图1A:多个域之间的scMMGAN架构映射,每个域由一对生成器和判别器组成;
- 图1B:除了判别器loss之外,每个域中还有两个附加loss;
- 图1C:关于数据几何(data geometry)的演示,通过correspondence loss指导对齐。在所描绘的空间中,两个域中的数据已被移位和旋转,但固有数据几何结构与扩散特征向量的值保持一致;
- 图1D:可反转(具有低重建损失)但不对齐几何表示(具有高对应损失)的不良映射(Bad mapping)和可反转且对齐几何表示的良好映射的假设说明。
通过计算原始域中每个点的扩散算子的特征向量 ϕ x 1 \phi_{x_{1}} ϕx1和映射到其他域后对应点的扩散算子的特征向量 ϕ G ( x 1 ) \phi_{G(x_{1})} ϕG(x1),我们可以直接比较第 i i i个特征向量从而对齐几何结构。然后,对应几何损失会对每个点的两个表示之间的L2距离进行惩罚: L c = ∣ ∣ ϕ x 1 i − ϕ G ( x 1 ) i ∣ ∣ 2 L_{c}=||\phi_{x_{1}}^{i}-\phi_{G(x_{1})}^{i}||_{2} Lc=∣∣ϕx1i−ϕG(x1)i∣∣2通过在scMMGAN框架中强制执行这种loss,我们确保在欠约束的GAN设置中保留数据的固有结构。
空间、scRNA-seq和蛋白质组学数据之间的映射
作为scMMGAN的初步验证,作者利用新开发的组织测序DBIT-seq中同时测量的多模态数据作为GroundTruth。作者将空间定位的scRNA-seq数据和空间定位的蛋白质数据视为两个独立的测量,并学习在它们之间进行映射。然后,利用它们是共同测量的这一事实,以及每个数据集中的一些列是相关的(对应的基因和蛋白质)来评估所学映射的准确性。图2显示了scMMGAN和基线模型在这些数据上的示例结果。结果表明scMMGAN对真实数据的建模效果最好。

- 图2:在DBIT序列数据上,显示了所示基因(Mpr1)的相应蛋白质组学和转录组学表达。绘制的x轴和y轴是直接从数据中获取的测量空间坐标(从蛋白质组学映射到RNA组学,并选择Mpr1基因的表达量作为定性评估结果)
RNA-seq和ATAC-seq之间的映射
接下来作者用成对的ATAC-seq和RNA-seq的测量设计了实验。作者使用的数据集来自于一个公开的人类血液数据集。图3显示了对结果进行的定性评估,第一栏是真实的RNA-seq的CLEC16A基因的分布,后面几栏是生成的相应RNA-seq的对应基因的分布。scMMGAN的输出与真实结果最匹配。对于其他模型,虽然它们在分布上有适当的激活量,但在point-wise的水平上,它们的排列是不准确的。
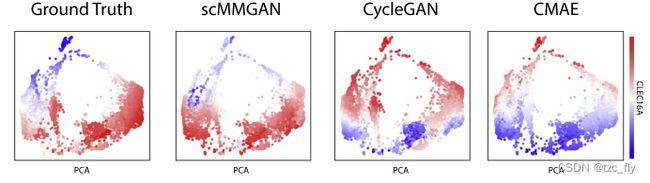
- 图3:实验比较结果,所示坐标来自主成分分析维度。
三阴性乳腺癌数据的整合
在这里,作者将scMMGAN应用于一个数据集,该数据集包括一个三阴性乳腺癌的人类异种移植模型(MDA-MB-231),其转录组的测量结果是由空间RNA-seq模态和缺乏空间信息的scRNA-seq模态共同完成的。虽然scRNA-seq和空间RNA-seq数据都是测量基因谱的转录组技术,因此它们的维度具有相同的意义,但这两个数据集不能按原样一起分析。在图4A中,可以看到在使用scMMGAN之前,这两个数据的分布是完全不重叠的。由于原始数据在联合空间中是完全可分离的,任何下游分析都只能发现两种模式之间的差异,而不能发现其中细胞之间的差异。对于使用两者信息的综合分析,需要scMMGAN的对齐输出(图4A)。
- 图4A:在原始数据中,空间RNA序列和scRNA序列不可直接比较,因为它们是完全可分离的。使用scMMGAN映射后,它们与下游分析对齐并可比较;
- 图4B:将空间RNA序列映射到scRNA序列,对生成的scRNA-seq进行聚类,然后通过x轴和y轴上测量的空间坐标绘制簇;可以发现对应簇与其标记基因相关。
- 图4C:从scRNA序列生成空间RNA序列数据,包括生成的空间坐标。坐标与前一图相同;
- 图4D:所有生成的scRNA-seq聚类后的簇映射到空间RNA序列空间;
局限
尽管GANs在映射分布方面很有用,但它们有一些关键的缺点。首先,由于对抗性损失,它们本身很难训练,这可能导致模型不稳定。这种不稳定性意味着模型可以在训练迭代中迅速从有效恶化到无效。此外,在基于每个映射方向的成对生成器的框架下,生成器的数量成倍增长。这意味着,对于大量的输入模态的对齐,网络必须做得很小,或者必须单独训练。