Colibri 片段化学空间的兴起
2007 年,Reymond 等人发布了 GDB-11 数据库,11 个重原子 2640 万个分子。时隔两年,再度发布的 GDB-13 数据库,13 个重原子 9.7 亿。2012 年,发布 GDB-17 数据库,激增到 17 个重原子 1660 亿!!
GDB 数据库是人类朝超大规模数据库进发的一个缩影。这种通过在边界约束下枚举出所有可能的有机分子,无疑人类探索化学空间的突出典范。
下图是一些知名枚举数据库的示意图。
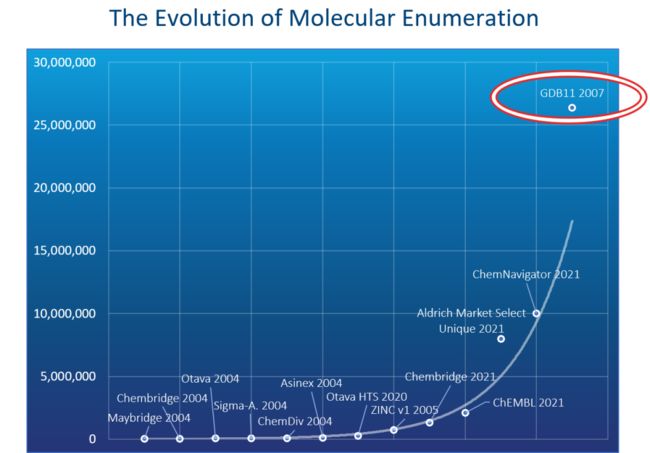
指数级的增长!!!

——枚举数据库的问题——
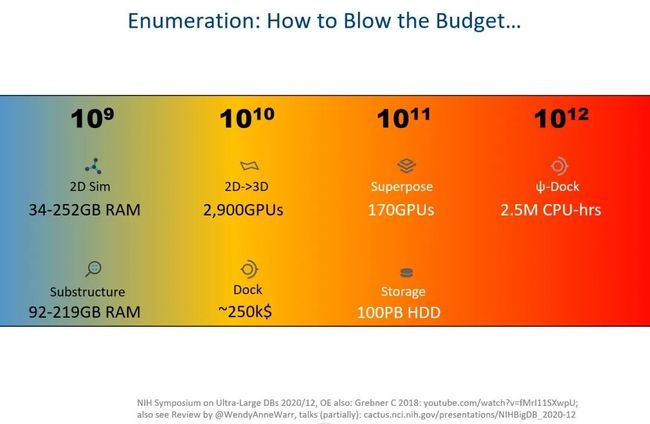
360-CPU cluster,100 000 CPU hours 才枚举生成了高达 1660 亿规模的 GDB-17 数据库。对于 1660 亿如此大的规模,就算以 zip 形式储存一维的 SMILES 格式,都需要大约 400 GB。做过虚拟筛选的人,应该很容易理解直接暴力对 1660 亿枚举数据库进行虚拟筛选,意味着什么!
下图是 NIH 研讨会中展示的枚举类数据库的操作成本和时间,如相似性检索、子结构检索、三维构象生成、分子对接、结构叠合和储存等等。
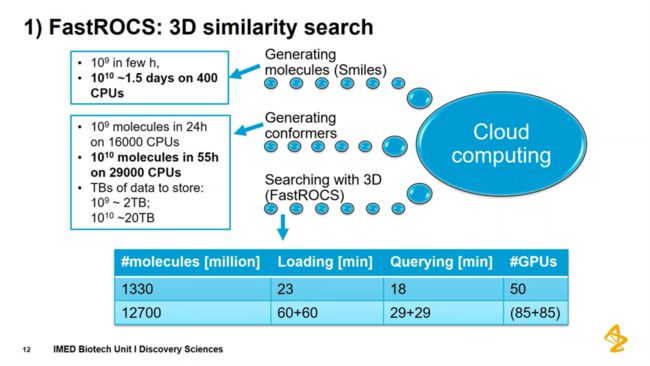
以 AstraZeneca 的实际项目为例,枚举出百亿级数据库,一维的 Smiles 格式 400 CPUs 1.5天。三维构象生成,29000 CPUs 55 小时。储存 20 TB。采用 FastROCS 进行三维相似性检索,1330 M,50 GPUs 加载 23min,查询 18 min。
和这种枚举类数据库相比,通过 infiniSee 检索百亿级片段化学空间,一台普普通通的办公电脑就可以完成,检索时间最快几秒钟,生成 10W 个解决方案也大多都在 30 min以内。天然之别!!
随后 2018年,AstraZeneca 对外公布完成内部 10 的 15 次方规模化学空间的构建。如今已经达到了 10 的 17 方。

可以预料,随着枚举数据库的激增,成本的增加没有尽头,效率也是极为低下。所以枚举类数据库通常的解决方式是检索部分数据库,而非全部,从而大大降低成本!!
除了成本和效率,这种枚举产生的虚拟数据库的可合成性也是大问题,如果通过筛选拿到了系列化合物,但是无法直接购买、甚至难以合成或者无法合成,那这个化合物就没有意义。
对于枚举数据库,常规做法是首先对化学空间进行均匀采样,然后对采样后的数据库进行虚拟筛选等操作,找到合适的化学型后,再针对该化学型周围的化学空间进行详细采用,如此进行迭代,如化学空间加速药物发现 @重复迭代。
很明显,这种方式受采样质量的影响,会漏掉很多有价值的数据,并非解决良策。能否高效率、短时间完整的遍历化学空间?直接进行相似性检索、虚拟筛选和子结构检索?
——化学空间的崛起——
预料到枚举数据库的尽头,大型制药公司开始基于分子砌块和化学反应构建片段化学空间。下图是全球化学空间示意图,有 BiosolveIT 标志的即为 Colibri 片段化学空间。
片段化学空间和枚举数据库最大的区别就是枚举类数据库都是以完整的化合物储存的,而片段化学空间只有分子砌块和分子砌块间的连接方式。所以超大规模枚举类数据库动则需要 PB 级别的储存(千亿),而片段化学空间一台最普通办公电脑就可以。
下方是片段组合的示意图,在进行片段化学空间的检索,如相似性检索、子结构检索和化学空间筛选时,分子砌块通过对应的连接方式即时生成完整的化合物。效率远远超过枚举类数据库!!!!

枚举类数据库和片段化学空间都有各自的优点儿。对于化学空间,只要有明确验证的化学反应和高质量高的分子砌块,就可以构建高质量可合成的片段化学空间。
对于可合成性,以四大制药公司的内部数据和 Merck 高达 10 的 20 次方的 MASSIVE 化学空间为例,合成率都在 80% 以上。Merck 内部数据表明在 12 个药物发现项目中,可合成性均在 80 % 以上,构建化学空间后,项目推进快了两倍,成本降低十倍!!而且内部分子砌块保证高 IP。这!就是构建片段化学空间的魅力!

略微可惜随着国外大型企业纷纷完成转型,成功完成企业内部化学空间的构建。但是中国制药企业还未有超大规模化学空间的报道。
商业可获得化学空间有四家 Enamine、WuXi、ChemSpace 和 OTAVA。可喜的是,药明康德构建了目前 80 亿的 GalaXi 化学空间,也算开了中国的先河。在 infiniSee 中检索 GalaXi,主观效率最高。
——化学空间的解决方案——
由于片段化学空间中并非完整的化合物,所以所有的检索方式都要重写。就算是基本的相似性检索、分子对接、子结构等等算法都需要重写。不过效果确实迥异。
以下方几个案例进行观察:
▎1. 相似性检索 - FTrees + Tanimote
化学空间最开始引起 Pfizer 和 Boehringer Ingelheim 等大型制药企业注意的就是相似性检索。在 2008 年,Pfizer 第一次系统的证明了 FTrees 在发现新化学型中的潜力。

相似性检索分为两种:
一是发现结构极为的化合物(Close-by,Tanimoto)。
二是发现结构有一定的差异性,但是可能具有相似生活活性的化合物(distant,FTrees)。
下图是 FTrees 示意图,绿点表明查询化合物,红点为结果,可以发现化学相似度变化较大时,生物活性并为有太大的降低。而且这点活性的丧失在后期结构修改时,可以很容易的改善。

FTrees 检索示意图
通常为发现全新化学型,会将 FTrees 相似度设置在 0.9 附近,以期发现具备同样相似生物活性,但是结构差异大的化合物。
目前,化学空间的检索算法较少,药企广泛使用的是 FTrees,目前集成在 infiniSee 中,同时内置五大化学空间,商业可获取的化学空间四个共 500 亿,还有一个基于文献和专利中的化合物和反应构建的 10 的 14 次方的化学空间,可以直接检索。
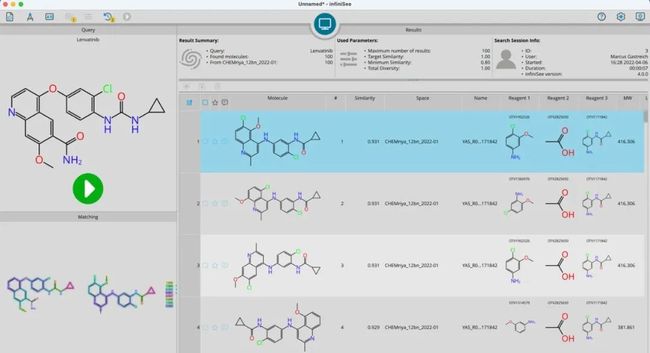
infiniSee 检索示意图(可后台回复任意文字联系小编试用)
但是!!FTrees 虽然有助于骨架跃迁,发现全新的结构,但是该检索形式是二维,假阳性较大,必须运用三维的方式进行后处理才可以得到合适的结果。
下图是一个常见的工作流,通过 infiniSee 进行二维的检索后,通过 ROCS 等进行三维形状过滤、约束对接、聚类分析和可视化检查。

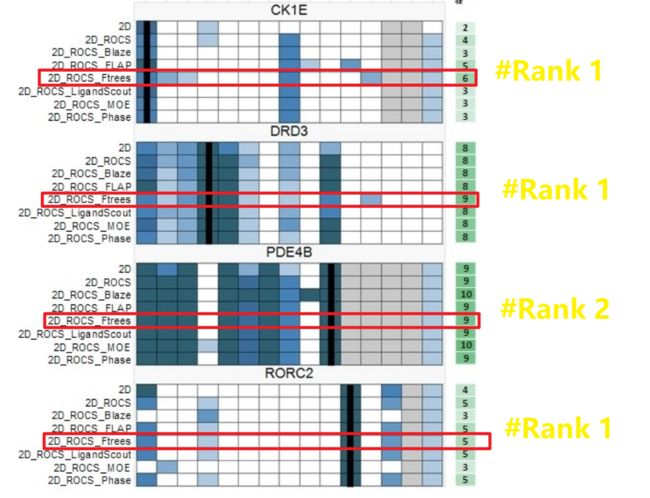
下图是辉瑞内部项目测试,在同样的数据库规模下,二维 FTrees 检索和三维方法的结合,可以实现最优的富集。
鉴于 FTrees 短时间可以实现百亿级以上化学空间的完整检索,可以远超于正常规模,理论富集率会更高。
基于相似性检索的自动化药物发现工作流可以参考:药物发现三种强大的自动化工作流。
▎2. 化学空间对接/筛选 - Chemical Space Docking
化学空间对接的核心理论就是先对接分子砌块,分子砌块基于化学反应进一步生长,最后形成完整的化合物。这也是目前可以实现百亿、万亿、兆亿或更大规模虚拟筛选的唯一方式。

以 Genentech 在今年 DrugSpace2022 大会上的报道为例,以 ROCK1 为例进行化学化学对接的概念验证(Chemical Space Docking)。
化学空间对接不仅可以轻松完成整个化学空间的探索,发现所有有可能的化学型。而且由于化学空间的特性,筛选出的化合物可以保证可合成性,药企根据内部的分子砌块,可高效的完成合成和后续检测。

最后的结构也是显而易见,选择的 69 个化合物中,有 27 个有效的化合物(阈值设置为 20 微摩尔),命中率高达 40 %。

这个技术未来肯定是大规模普及,其中的震撼,真的是懂得人才懂!!
对于规模越大的数据库,化学空间的魅力也就越大,下图是计算资源随着分子数据增加的二维图,化学空间对接在计算资源的优势显而易见。

▎3. 片段生长 - FastGrow
化学空间对接是针对化学空间中的所有化合物,有时候如果基于片段进行药物发现,确定初始片段片段后,可以直接基于初始片段进行后续生长。
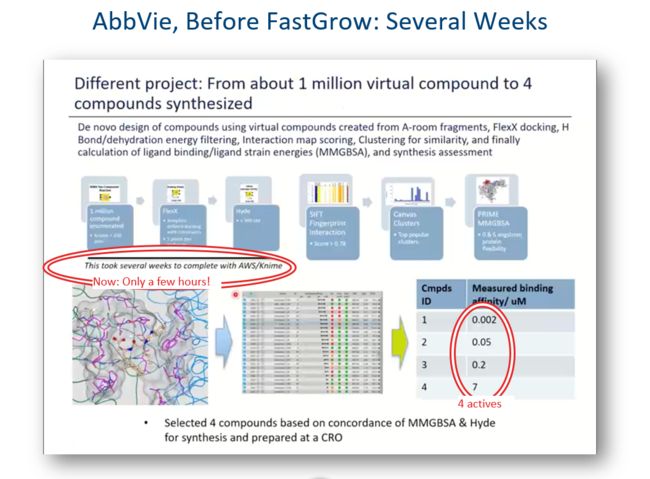
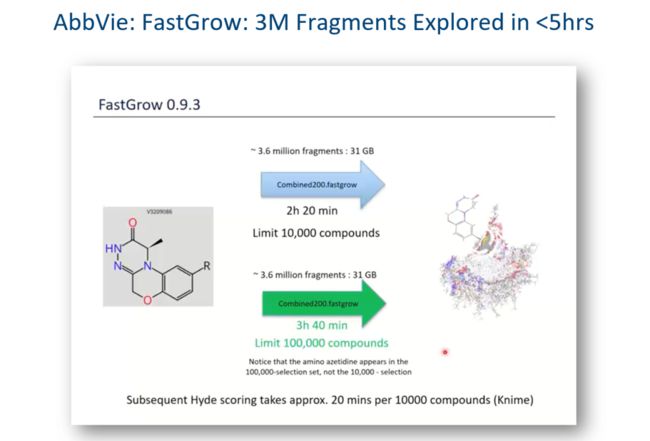
以 AbbVie 的案例为例,360 万的片段通过 FastGrow 需要多久?
AbbVie 的案例是以 PKC theta 为例,对于 FBDD 项目而言,拿到初始片段后,可以根据计算模拟筛选现有的片段库,根据化学可衍生部位进行片段生长以得到可能有效的药物。

具体可参考:艾伯维 | FastGrow 在类药性化合物设计和合成中的应用。
可以发现完整遍历 360 万的片段库,给出 1 万个结果只需要 2h 20min,给出 10W 个结果只需要 3h 40min。注意这里是 360W 片段库的完整遍历。而常规对接需要多长时间?!
完成片段生长后,无需对接,直接通过 HYDE 计算亲和力,然后进行过滤即可。

FastGrow 诞生前,原本需要几周完成的工作量,如今几个小时就可以轻松完成!!这对效率的提升不言而喻。