科研双十一,带你系统学习药物设计,快来看吧
利用高性能计算机来进行药物虚拟筛选已经被广泛应用,计算机辅助药物设计可以提高药物研发的成功率,降低研发成本,缩短研发周期,是目前创新药物研究的核心技术之一。随着医药大数据的积累和人工智能技术的发展,运用AI技术并结合大数据的精准药物设计也不断推动着创新药物的发展。在新型冠状病毒的治疗方案中,通过一系列计算机辅助药物生物计算的方法发现一大类药物分子可以有效阻止新冠病毒的侵染,为治疗新冠提供了新思路。
分子动力学模拟是分子模拟中最接近实验条件的模拟方法,能够从原子层面给出体系的微观演变过程,直观的展示实验现象发生的机理与规律,促使我们的研究向着更高效、更经济、更有预见性的方向发展。分子动力学可以解决和研究DNA的折叠和性质、蛋白与配体的识别机制、跨膜蛋白的工作机理、蛋白酶与底物的反应、蛋白与蛋白的耦合、比较野生型与突变蛋白的不同特性、蛋白折叠的机制问题(控制温度,使蛋白自行折叠和去折叠)等。
专题一:CADD蛋白结构分析、虚拟筛选、分子对接(蛋白-蛋白、蛋白-多肽等)
2022年11月19日-11月20日 在线直播(授课2天)
2022年11月26日-11月27日 在线直播(授课2天)
2022年12月03日-12月04日 在线直播(授课2天)
专题二:AMBER分子动力学能量优化与分析、结合自由能计算专题
2022年12月10日-12月11日 在线直播(授课2天)
2022年12月17日-12月18日 在线直播(授课2天)
专题三: GROMACS分子动力学蛋白模拟、药物开发溶剂筛选专题
2022年12月24日-12月25日 在线直播(授课2天)
2022年12月31日 在线直播(授课1天)
专题四:AIDD人工智能(机器学习与深度学习)药物发现专题
2022年12月10日-12月12日 在线直播(授课3天)
2022年12月17日-12月18日 在线直播(授课2天)
- 年底大赠送:凡报名价格高的专题课程,可任选一个比所报课程价格低的课程免费参加;
- 凡报名学员将获得本次培训书本(或电子)教材以便提前预习及随堂电子资料;
- 凡报名学员将获得课程相关Windows版本软件安装指导(一年有效期);
- 提供无限次回放视频:凡报名学员课程结束后可获得所学专题课程无限次回放视频;
- 价格优惠:(优惠活动最终解释权归主办方)
- 早鸟优惠:所有专题课程2022年11月1日前报名缴费均立减200元
- 转发优惠:转发本文/海报至朋友圈12小时,优惠100元/人(朋友圈不设屏蔽,截图联系工作人员确认)
- 凡老学员推荐的新报名者可享受额外优惠
- 老学员优惠:老学员参加任一新课程立减400元(与1)2)不叠加)
- 学员提出的各自遇到的问题在课程结束后可以长期得到老师的解答与指导;
- 参加培训并通过考试的学员,可以获得:北京软研国际信息技术研究院培训中心颁发的《计算机辅助药物设计应用工程师》专业技能结业证书;
专题一:CADD蛋白结构分析、虚拟筛选、分子对接(蛋白-蛋白、蛋白-多肽)
| 时间 |
课程名称 |
课程内容 |
| 第一天 上午 |
生物分子互作基础 |
1.生物分子相互作用研究方法 1.1蛋白-小分子、蛋白-蛋白相互作用原理 1.2 分子对接研究生物分子相互作用 1.3 蛋白蛋白对接研究分子相互作用 |
| 蛋白数据库 |
1. PDB 数据库介绍 1.1 PDB蛋白数据库功能 1.2 PDB蛋白数据可获取资源 1.3 PDB蛋白数据库对药物研发的重要性 2.PDB 数据库的使用 2.1 靶点蛋白结构类型、数据解读及下载 2.2 靶点蛋白结构序列下载 2.3 靶点蛋白背景分析 2.4 相关数据资源获取途径 2.4 批量下载蛋白晶体结构 |
|
| 第一天 下午 |
蛋白结构分析 |
1. Pymol 软件介绍 1.1 软件安装及初始设置 1.2 基本知识介绍(如氢键等) 2.Pymol 软件使用 2.1蛋白小分子相互作用图解 2.2 蛋白蛋白相互作用图解 2.3 蛋白及小分子表面图、静电势表示 2.4蛋白及小分子结构叠加及比对 2.5绘相互作用力 2.6 Pymol动画制作 实例讲解与练习: (1)尼洛替尼与靶点的相互作用,列出相互作用的氨基酸,并导出结合模式图 (2)制作结合口袋表面图 (3)Bcr/Abl靶点的PDB结构叠合 (4)制作蛋白相互作用动画 (5)针对ACE2和新冠病毒Spike的蛋白晶体复合物,制作蛋白-蛋白相互作用 |
| 第二天上午 |
同源建模 |
1.同源建模原理介绍 1.1 同源建模的功能及使用场景 1.2 同源建模的方法 2. Swiss-Model 同源建模; 2.1 同源蛋白的搜索(blast等方法) 2.2 蛋白序列比对 2.3 蛋白模板选择 2.4 蛋白模型搭建 2.5 模型评价(蛋白拉曼图) 2.6 蛋白模型优化 实例讲解与练习:用2019-nCoV spike蛋白序列建模,根据相应参数和方法评价模型 |
| 第二天下午 |
小分子构建 |
1. ChemDraw软件介绍 1.1小分子结构构建 1.2 小分子理化性质(如分子量、clogP等)计算 实例讲解与练习:分别构建大环、氨基酸、DNA、RNA等分子 |
| 小分子化合物库 |
1. 小分子数据库 1.1 DrugBank、ZINC、ChEMBL等数据库介绍及使用 1.2 天然产物、中药成分数据库介绍及使用 |
|
| 第三天上午 |
生物分子相互作用Ⅰ |
分子对接原理及对接软件介绍 2. 分子对接软件(Autodock) 使用 2.1半柔性对接 2.1.1 小分子配体优化准备 2.1.2 蛋白受体优化及坐标文件准备 2.1.3 蛋白受体格点计算 2.1.4 半柔性对接计算 2.2对接结果评价 2.2.1 晶体结构构象进行对比 2.2.2 能量角度评价对接结果 2.2.3 聚类分析评价对接结果 2.2.4 最优结合构象的选择 2.2.5 已知活性化合物对接结果比较 实例讲解与练习:激酶Bcr/Abl靶点抑制剂的半柔性对接 |
| 第三天下午 |
生物分子相互作用II |
2.3柔性对接 2.3.1 小分子配体优化准备 2.3.2 蛋白受体优化及坐标文件准备 2.3.3 蛋白受体格点计算 2.3.4 柔性对接计算及结果评价 2.3.6 半柔性对接与柔性对接比较与选择 实例讲解与练习:Bcr/Abl靶点抑制剂的柔性对接 |
| 虚拟筛选 |
3. 分子对接用于虚拟筛选(Autodock) 3.1 虚拟筛选定义、流程构建及演示 3.2 靶点蛋白选择、化合物库获取 3.3虚拟筛选 3.4 结果分析(打分值、能量及相互作用分析) 实例讲解与练习:Bcr/Abl靶点抑制剂的虚拟筛选 |
|
| 小分子格式转换 |
1.1 openbabel软件介绍 1.2 小分子结构类型 1.3 小分子结构类型转换 |
|
| 答疑 |
针对前三天学习问题的答疑 |
|
| 第四天上午 |
拓展对接使用场景(上) |
1.蛋白-蛋白大分子对接 1.1蛋白-蛋白对接的应用场景 1.2相关程序的介绍 1.3 受体和配体蛋白前期优化准备 1.4 载入受体和配体分子 1.5蛋白蛋白相互作用对接位点设定 1.6蛋白蛋白对接结果分析与解读 实例讲解与练习:新冠病毒Spike蛋白及宿主蛋白ACE2的对接
2.蛋白-多肽对接 2.1蛋白-多肽相互作用简介 2.2蛋白-多肽分子预处理 2.3蛋白-多肽分子对接 2.4对接结果展示与分析 实例讲解与练习:新冠靶点3CL与多肽/类多肽抑制剂的对接 3.含金属离子的蛋白靶点与小分子对接
3.1 金属酶蛋白-配体的相互作用介绍 3.2相关蛋白及配体分子的收集与预处理 3.3金属离子的处理与准备 3.4金属辅酶蛋白-配体的对接 3.5对接结果展示与分析
|
| 第四天下午 |
拓展对接使用场景(下) |
4.小分子与小分子对接 4.1小分子-小分子相互作用简介 4.2小分子结构预处理 4.3小分子-小分子对接 4.4对接结果展示与分析 实例讲解与练习:环糊精与药物小分子的对接 5.核酸-小分子对接 5.1核酸-小分子的应用场景 5.2核酸-小分子相互作用简介 5.3核酸-小分子的预处理
5.4核酸-小分子对接 5.5相关结果的展示与分析 实例讲解与练习:DNA G-四链体和配体分子对接 6.共价对接 6.1共价对接的原理及应用场景 6.2蛋白和共价结合配体的预处理 6.3药物分子与靶蛋白的共价对接 6.6相关结果的展示与分析 实例讲解与练习:激酶靶点EGFR抑制剂的共价对接 |
| 第五天上午 |
基于碎片药物设计 |
1.1基于碎片的药物设计与发现 1.2 基于碎片化合物库构建 1.2.1 骨架替换 1.2.2 碎片连接 1.2.3 碎片生长 1.3 基于药效团的化合物库生成 1.4 基于蛋白结合口袋的化合物库生成 1.5 基于分子描述符的化合物库生成 1.6 基于BREED规则的化合物库构建 1.7 基于碎片的化合物库筛选 实例讲解与练习:基于片段的Bcr/Abl靶点抑制剂优化与改造 |
| 第五天下午 |
构效关系分析 |
1. 3D-QSAR模型构建(Sybyl软件) 1.1 小分子构建 1.2 创建小分子数据库 1.3 小分子加电荷及能量优化 1.4 分子活性构象确定及叠合 1.5 创建3D-QSAR模型 1.6 CoMFA和CoMSIA模型构建 1.7 测试集验证模型 1.8 模型参数分析 1.9 模型等势图分析 1.10 3D-QSAR模型指导药物设计 实例讲解与练习:激酶靶点Bcr/Abl抑制剂的构效关系模型构建与活性预测 |
| 第六天全天 |
分子动力学模拟 |
1. 分子动力学简介(GROMACS软件) 1.1分子动力学基本原理 1.2 Linux 系统介绍 1.3 分子动力学软件介绍(Gromacs) 2. Gromacs 进行分子动力学模拟 2.1 配体分子的处理 2.2 蛋白结构的处理 2.3 修改蛋白坐标文件 2.4修改拓扑文件 2.5构建盒子并放入溶剂 2.6平衡系统电荷 2.7能量最小化 2.8 NVT平衡 2.9 NPT平衡 2.10 产出动力学模拟 3. 分子动力学结果分析 3.1轨迹文件观察 3.2能量数据作图 3.3 轨迹修正处理 3.4 回旋半径分析 3.5 计算蛋白构象的RMSD 变化 3.6计算原子位置的RMSF变化 3.7 蛋白配体构象聚类 3.8蛋白配体相互作用氢键分析 3.9 蛋白配体相互作用能分析 实例讲解与练习: (1)水中的溶菌酶纯蛋白模拟 (2)T4溶菌酶及配体复合物模拟 |
| 答疑 |
针对后三天学习问题的答疑 |
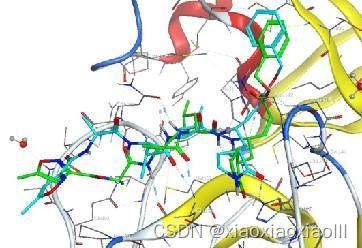
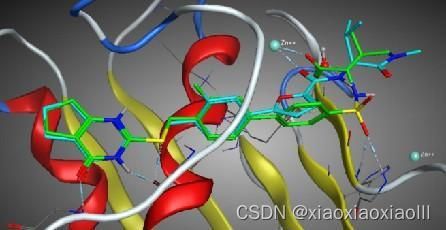
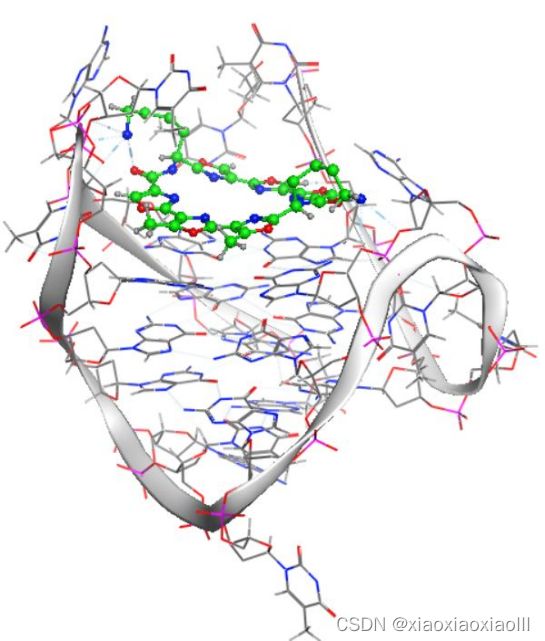
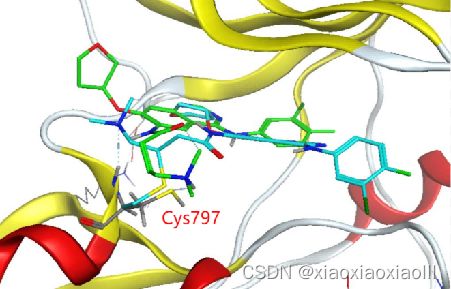
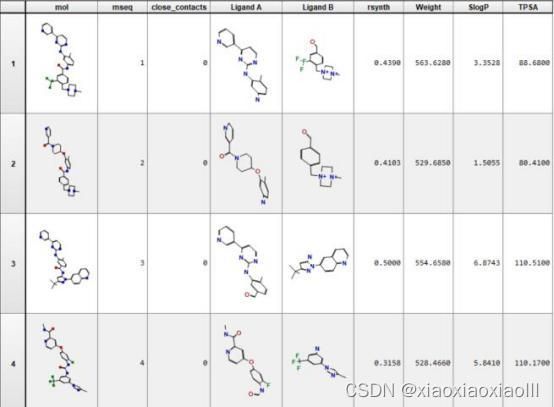
专题二:AMBER分子动力学能量优化与分析、结合自由能计算专题
| 第一天 |
||
| 时间 |
课时内容 |
主要知识点 |
| AM 9:00~9:50 |
教学目标:了解本方向内容、理论基础、研究意义。 |
|
| AM 10:00~10:50 |
教学目标:掌握数值计算平台,熟悉计算机语言,能够使用vim编辑器简单编辑文件。 |
|
| AM 11:00~12:00 |
||
| PM 14:00~14:50 |
教学目标:了解Amber软件历史发展,熟悉安装环境,支撑环境编译。 |
|
| PM 15:00~15:50 |
||
| PM 16:00~17:00 |
||
| 第二天 |
||
| 时间 |
课时内容 |
主要知识点 |
| AM 9:00~9:50 |
教学目标:如何确立研究对象,熟悉蛋白数据库的使用,如何对 研究对象建模。 |
|
| AM 10:00~10:50 |
教学目标:熟悉模型预处理流程,掌握输入文件的编写,能够独立完成体系动力学之前的准备工作。 |
|
| AM 11:00~12:00 |
案例实践:
|
|
| PM 14:00~14:50 |
教学目标:分子动力学流程,AMBER软件动力学原则,完成分子动力学模拟的操作练习。 |
案例实践:
|
| PM 15:00~15:50 |
||
| AM 16:00~17:00 |
||
| 第三天 |
||
| 时间 |
课时内容 |
主要知识点 |
| AM 9:00~9:50 |
教学目标:熟悉结合自由能计算的意义、MMPBSA方法以及流程。 |
|
| AM 10:00~10:50 |
案例实践:
|
|
| AM 11:00~12:00 |
||
| PM 14:00~14:50 |
教学目标:熟悉可视化软件获取渠道、软件安装以及基本使用,采用可视化软件辅助科研工作。 |
|
| PM 15:00~15:50 |
教学目标:从动力学模拟出的构象出发,洞悉构象转变,解释实验想象,预测实验结果 |
|
| AM 16:00~17:00 |
|
|
| 第四天 |
||
| 时间 |
课时内容 |
主要知识点 |
| AM 9:00~9:50 |
教学目标:从能量角度出发,分析分子间、残基间、重要基团间相互作用机理,对实验提供理论指导 |
|
| AM 10:00~10:50 |
||
| AM 11:00~12:00 |
||
| PM 14:00~14:50 |
教学目标:引导初学者了解本方向中经典工作,复现工作中重要分析手段,加深同学对本方向的理解。 |
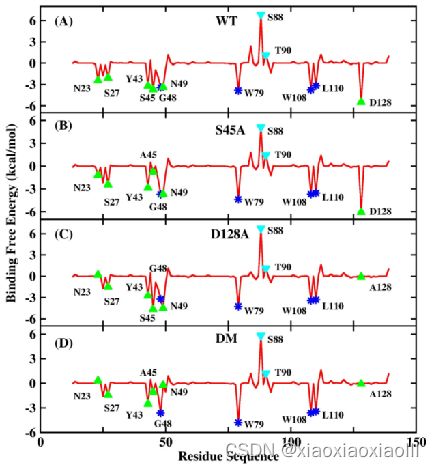
(a) 丙氨酸扫面预测热点氨基酸 (b) 残基接触(contact)分析 (c) 温度因子分析(B-facter)分析 (d) 溶剂可及表面积分析 (e) 伞状采样动力学再现解离机制
|
| PM 15:00~15:50 |
||
| PM 16:00~17:00 |
||
部分案例图示:

图一:残基分解
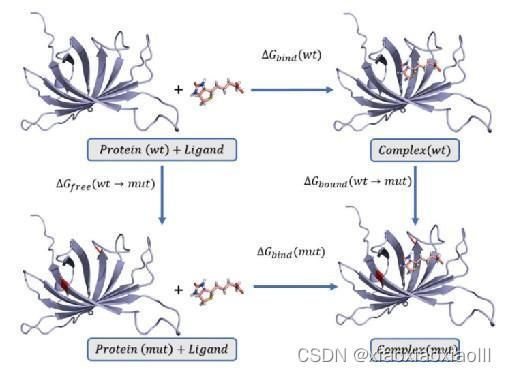
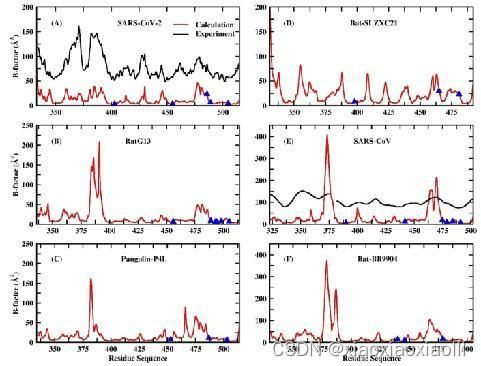
图二:热力学积分计算相对结合自由能 图三:温度因子分析(B-facter)分析
专题三:GROMACS分子动力学蛋白模拟、药物开发溶剂筛选专题
| 课程名称 |
内 容 |
| 基础理论 |
1 分子模拟基础理论 1.1 统计力学理论概述 1.2 主要算法介绍:最速下降法、共轭梯度法、有限差分法 1.3 力场、力场类型、参数和分类:AMBER、CHARMM、MMX、CVFF、OPLS 1.4 基础知识:积分迭代器、积分步长选取、温度控制、压力控制、周期性边界条件 1.5 模拟基本流程:能量最小化、NVE弛豫、NVT控温、NPT控压、MD平衡模拟 1.6 计算化学基本概念:范德华表面、分子表面、接触表面、溶剂可及表面、势能面 |
| 程序入门 |
2 GROMACS程序入门——学会编译方法,安装自己的GROMACS可执行程序,并运行一个例子。 2.1 版本/安装/运行 •Linux入门操作及方法 •并行介绍和环境搭建 •win版、linux版编译安装及运行 •win版下使用linux系统编译安装及运行GROMACS程序 2.2 各种文件介绍:PDB、GRO、TOP/ITP、XVG、MDP 2.3 力场概念、分类及力场参数修改:——探究力场具体形式,为以后创建自己体系做准备,以OPLS为例,力场的各种参数说明及修改 |
| 生物体系建模与TOP文件构建 |
3 生物体系建模——掌握不同体系快速搭建方法,使学员具有扎实的建模基础 3.1 辅助工具软件:Packmol、GaussView、vmd、Grace等 3.2 模型建模/TOP文件的生成 生物小分子PDB构建、生物小分子模型及原子类型定义、结构调整(键长、键角、二面角)、生物小分子top结构构建、itp文件建立、拓扑文件生成工具 3.3 不同简单生物体系的建模 3.4 构建一个简单的生物分子体系模型并运行 3.5 蛋白质、核酸、多肽、溶剂等复杂体系构建 |
| 建模结果分析 |
4 模拟结果分析——掌握不同生物体系所需分析方法,生成拓扑结构和坐标文件 4.1 模拟轨迹分析:trajectory,sasa,rdf,freevolume等 4.2 生成拓扑结构和坐标文件:editconf,genconf,pdb2gmx等 4.3 模拟能量分析:energy,enemat等 4.4 系统动态结构分析:cluster,confrms,midist等 4.5 空间分布性质:gyrate,msd,rdf,traj等 4.6 分子结构分析:hbond,order,principal,spol等 4.7 静电作用分析:dielectric,dipoles,potential等 |
| 生物体系建模实践 |
5 水溶性蛋白质和配体作用分子动力学模拟 5.1 配体分子的处理 5.2 蛋白结构的处理 5.3 修改蛋白坐标文件 5.4 修改拓扑文件 5.5 构建盒子并放入溶剂 5.6 平衡系统电荷 5.7 能量最小化 5.8 NVT平衡 5.9 NPT平衡 5.10 模拟结果取样 6 分子动力学结果分析 6.1 轨迹文件观察 6.2 能量数据作图 6.3 计算斜方差 6.4 测量回旋半径 6.5 计算结构的RMSD值 6.6 计算原子位置的根均方波动 6.7 计算模拟过程中分子间的氢键的数目、距离或角度 7.1 磷脂分子结构、双分子层建模 7.2 模拟水通道(蛋白、多肽等) 7.3 插入溶剂分子 7.4 模拟系统达平衡 7.5 分析溶剂分子扩散速率 7.6 分子间的氢键的数目、距离或角度 |
| 高级进阶模拟分子自由能计算实践 |
8 蛋白质结合自由能计算(伞形采样法为例) 8.1 创建一系列反应路径分子构型 8.2 提取模拟间隔质心轨迹 8.3 模拟每个构型的伞形采样 8.4 柱状图分析计算结合自由能 8.5 模拟结果讨论
9 药物分子开发溶剂筛选 9.1 介绍热力学积分方法 9.2 构建药物分子模型 9.3 建模药物分子溶解在水相,油相和醇相 9.4 计算不同相态下的分配系数 9.5 快速预测药物分配系数方法 |
| SCI论文复献 |
根据学员需求,回答相关SCI文献中模拟的问题 |
专题四:AIDD人工智能(机器学习与深度学习)辅助药物发现专题
| 时间 |
课程内容 |
课程内容 |
| 第一天上午 |
计算机辅助药物分子设计 |
1.计算机辅助药物设计(CADD)简介 2.CADD的基本方法 2.1分子对接 2.2药效团 2.3 QSAR和QSPR 2.4 各类药学研究数据库的介绍 |
| 第一天下午 |
Anaconda3的安装配置 |
3.Anaconda3 3.1 Pandas 3.2 NumPy 3.3 RDKit 3.4 scikit-learn 3.5 Pytorch 3.6 Tensorflow 3.7 DeepChem 3.8 XGBoost |
| 第二天上午 |
AIDD简介 ——分类和回归任务 |
1.分类模型的构建与应用 1.1逻辑回归算法原理 1.2朴素贝叶斯算法原理 1.3k最近邻算法原理 1.4支持向量机算法原理 1.5随机森林算法原理 1.6梯度提升算法原理 2. 分子特征介绍 2.1 分子描述符 2.2 分子指纹 2.3分子图 |
| 第二天下午 |
基于浅层机器学习的药物发现(目标:引导学员自行实现基于其他三种算法如KNN,SVM,XGBoost的毒性预测模型,并用于小分子化合物的毒性预测) |
3.模型评估方法 3.1交叉验证 3.2外部验证 3.3分类模型的常用评价指标 3.4混淆矩阵 3.5准确率 3.6敏感性 3.7特异性 4.参数优化与模型选择 4.1超参数优化4.2 模型选择的标准 模型实例讲解与练习(文献复现):
|
| 第三天上午 |
基于浅层学习的药物发现——回归任务 |
1.回归模型的构建与应用 2.回归模型的常用评价指标 2.1 MSE 2.2 MAE 2.3 R2 3.模型选择 3.1超参数优化 3.2最优模型选择 |
| 第三天下午 |
基于浅层学习分类的虚拟筛选(目标:引导学员自行实现基于其他三种算法的pIC50值预测模型,并用于小分子化合物pIC50值的预测) |
模型实例讲解与练习(文献复现):
|
| 第四天 |
基于深度学习的药物发现 |
1.深度学习的发展历程 1.1深度学习在药物开发中的应用 1.2 DNN 1.3 GCN 1.4 GAT 1.5 KGCN 1.6 FP-GNN 2.深度神经网络的常用框架介绍 2.1 PyTorch 2.2 TensorFlow 3.DEEPCHEM介绍与使用 模型实例讲解与练习(文献复现):
|
| 第五天 |
使用DNN,GCN,GAT等主流深度学习模型进行实操 |
实例讲解与练习(文献复现):
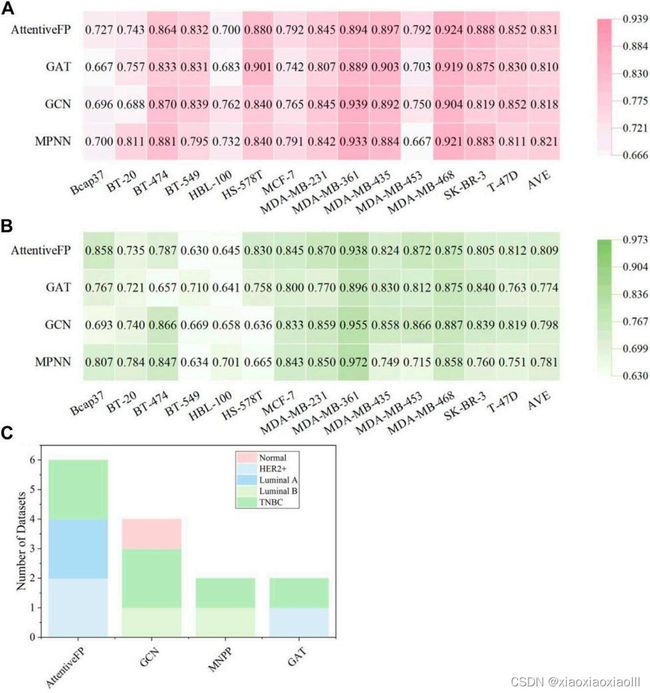
基于图模型的F1分数(A);基于图模型的AUC结果(B);基于分子图的不同亚型细胞的最优模型(C) |