宏病毒组富集技术详解
宏病毒组富集技术详解
病毒可以说是地球上最大的生物实体之一,但是由于他们具有形态小,遗传物质进化快等的特点,因而不能像细菌、古菌等微生物一样有系统的发育标记,这为其鉴别增加了一定的难度。此外,对于病毒与宿主之间相互关系的研究,也被限制在了可培养的病毒-宿主系统中,这使得对其研究十分困难。
目前对病毒组的了解主要是通过以下几个技术的结合,其中包含了传统的病毒培养技术、高分辨率显微镜技术,PCR技术及高通量宏基因组测序分析技术(表1)。目前,通过采用与肠道细菌组的研究相类似的方法,对肠道病毒组中相关复杂病毒群落进行分析,已摆脱对传统培养及显微镜的依赖,进而转向新一代宏基因组测序技术。


(表1)宏病毒组技术汇总
基于培养的技术(包括利用特定的细菌宿主从环境中分离噬菌体)使人类对病毒有了初步的认知,并被视为继续探索人类肠道病毒组的重要工具。事实上,目前对噬菌体与宿主的互作关系、噬菌体的功能和生态学的认知,在很大程度上基于长期以来细致的体外研究方法,如通过传统菌斑测定等经典病毒学方法进行噬菌体分离。由于环境中超过99%的细菌种类不可培养,使得这种技术受到了很大程度的限制(Amann, et al, 1995; Rappé, et al, 2003);但目前正在进行的研究大幅增加了可培养的代表性肠道微生物群落(Rajilic ́-Stojanovic ́ and de Vos, 2014)。由于目前的体外观察结果多在高度控制的、标准化的、基本上为人工营造的实验室条件下(essentially artificial laboratory conditions)产生,因此其结果究竟在多大程度上反映了体内的情况尚不确定。尽管一些体外模型可以复制肠道病毒组内的某些特征,但这些体外模型的可行性及代表性仍有待商榷。
透射电子显微镜(transmission electron microscopy, TEM)等高分辨率显微镜分析技术是研究噬菌体结构和识别不同形态噬菌体类型的必要手段。在其之前,病毒的结构和形态几乎是完全未知的,只有通过观察某些能够引起原发疾病的无细胞组织提取物,才能知道病毒的存在。此外电子显微镜还提供了基于噬菌体结构和形态对人类肠道病毒组组成的早期认识,这是病毒分类系统中的重要参数。这些研究表明,尾状噬菌体(Caudovirales)普遍存在(Flewett, et al, 1974; Letarov and Kulikov, 2009);近期基于宏基因组的数据集也证实和扩展了其观察结果,表明Caudovirales目(长尾噬菌体科Siphoviridae,肌尾噬菌体科 Myoviridae, 短尾噬菌体科 Podoviridae)中的有尾,双链DNA病毒与无尾单链DNA病毒占据比例优势(Kim et al, 2011)。
测序技术和生物信息学方法的快速进展使研究者能够更深入地了解病毒群落的结构和功能。利用过滤和密度梯度超离心技术从样本中分离病毒样颗粒(virus-like particles, VLPs),提取和扩增病毒核酸,并进行测序(Thurber, et al, 2009 and Reyes, et al, 2012)。这种方法为病毒组研究带来了前所未有的机遇,并深入了对病毒组的组成及功能的探索(Breitbart, et al, 2003; Reyes, et al, 2011; Kim, et al, 2011; Minot, et al, 2011), 但由于不同的方法对VLPs的富集有较大影响,从而影响最终结果。因此,根据研究目的,并对VLPs富集方法进行正确的选择至关重要。后文将对目前常用的VLPs富集方法的比较,进行探讨。
宏基因组技术在病毒组研究中的优势及挑战
与细菌组成部分的研究一致,宏基因组学方法现已成为病毒生态学研究的主体,并有望开创病毒生态基因组学的新时代,这类似于微生物生态学最近的复兴。然而,将宏基因组技术应用于病毒组学研究中时所产生的结果及对结果的解读仍面临许多挑战。如在提取VLPs的过程中,取样时存在的游离的、可活跃复制的噬菌体颗粒成为分析的重点,因而限制了对部分静态噬菌体颗粒的信息的获取。此外,由于提取的DNA水平较低,实施扩增程序可能会漏掉某些噬菌体类型(Angly, et al, 2006),这会使获得的病毒组的信息产生偏差。然而,对于群落级病毒组学调查而言,宏基因组学方法最重要的问题在于不能提供有效的宿主范围信息。尽管独立于培养的方法减轻了繁杂的实验工作,并能获得更大量的信息,但研究者必须使用其他生物信息学方法来间接推断病毒的宿主范围信息。
现阶段对肠道噬菌体的功能和系统发育构成的认知主要基于比对技术,如利用核苷酸和氨基酸水平上序列之间的同源性,进行Blast系列算法比对等(Altschul, et al, 1990)。这些方法也凸显了噬菌体基因组编码的大量新的基因内容。然而目前的研究发现,多数噬菌体基因与现有数据库条目缺乏同源性(Breitbart, et al, 2003; Reyes, et al, 2010; Minot, et al, 2011),这为了解病毒群落的基本结构和功能的研究带来了重大挑战。此外,由于缺乏已经明确感染关键细菌的噬菌体参考基因组序列,更使得对病毒组的研究困难重重。缺乏感染拟杆菌(Bacteroides spp.)的噬菌体的基因组数据很好地证明这一问题,其相关的噬菌体与炎症介导的疾病(Wexler 2007; Mazmanian, et al, 2008)的发生和保护有关。据预测,目前所具有的全球噬菌体序列数据库占实际噬菌体的比例不到0.001%(Kazankov, et al, 2014)。相比之下,基于宏基因组测序对肠道微生物的研究,已经大大受益于从各种生物和环境下日益完善的基因组序列数据集。
由于在病毒中缺乏一个类似于细菌中16S rRNA的系统发育锚点基因,使病毒组的进化和生态学研究也受到了阻碍。尽管保守的病毒基因确实存在,如噬菌体中的末端酶基因(terminase gene)(Casjens, 2003),但噬菌体基因组呈现的显著嵌合现象足以掩盖任何强大的系统发育信号。
直接从样本中(如粪便)提取的DNA,基本包含了当下环境中样本的全部宏基因组信息,其中有1%-17%为病毒DNA(Qin, et al, 2010; Minot, et al, 2011)。为此,样本整体的DNA信息被视为分析噬菌体群落的宝贵信息资源,特别是其与VLPs衍生的数据集放在一起时,为研究生态系统中噬菌体的组成提供了更为准确的宏观描述。尽管有这些潜在的优势,但无论是从VLPs,还是宏基因组中获得的数据,都为宏病毒组的分析提出了诸多挑战:如何更好的区分病毒和细菌的reads、如何将reads匹配给不同的基因组以及如何将宿主物种与已知的噬菌体匹配等。目前通过更完善的病毒组数据库,更优化的生物信息流程,已逐步解决所面临的一些挑战(详见本书生物信息部分)。
综上所述,在宏病毒组研究中所面临的挑战并非仅来源于病毒的多样性,完善病毒的参考基因组,明确其宿主范围及功能同样势在必行。
VLPs常用富集方法及优劣比较
由于绝大部分病毒的基因组太小,因此在微生物群落的总 DNA 中,来自病毒组的DNA所占比例非常小,尤其是其在低水平存在的情况下更难获得,但由于病毒的大小和特殊的形态,更易于通过一些物理或化学手段将其进行富集、分离,因而病毒较小的基因组更有利于使用生物信息技术进行全基因组组装。综上,高通量测序的手段更适用于病毒组的研究。
在宏病毒的研究中,VLPs生物富集和纯化是至关重要的。样本中VLPs纯化方法大致为:先用缓冲液制备样本匀浆液,然后根据不同微生物个体大小或密度重量的差异,用滤膜过滤或CSCl梯度密度离心的方式去除宿主细胞和细菌等污染,以达到富集病毒颗粒的目的。不同的化学试剂与不同的滤膜,对颗粒的富集及后续的数据结果,都会有一定的影响。
不同实验条件(滤膜,离心,化学处理等)对病毒颗粒富集的影
离心,过滤和氯仿处理是富集病毒颗粒的常用方法。对具有与小型细菌相似的物理尺寸的巨型病毒的报道越来越多,这使得病毒颗粒的富集越发具有挑战。
在过滤之前,离心已广泛用于病毒学研究中,而且对病毒组量化的影响较小。高速离心是病毒组颗粒富集中常用的方法,以17000 g离心3分钟几乎可去除所有细菌,但同时也降低了巨型病毒的数量,如mimivirus。尽管细菌中核糖体的大小(20–30nm)与许多病毒的大小相似(表2),但是17000 g离心仍有效去除了约99%的rRNA,主要原因可能是核糖体的密度比病毒颗粒更高,导致其更有效的沉淀;或者,绝大部分rRNA可能仍已附着于较大的细菌细胞,并随之一起沉淀。
在噬菌体颗粒富集中,对于滤膜的选择,主要包含0.22μm与0.45μm。那么如何选择合适滤膜,对多数的研究者造成不小的困扰。由表2可以看出,常见代表性病毒的大小由17nm到1000nm不等,而常见的噬菌体体积小于125nm,由此可见,滤膜大小的选择,取决于研究的主体是否包含巨型病毒,以及对病毒组的活性需求。例如,由于肠道病毒组中绝大部分由噬菌体组成,为了避免细菌的污染,在研究肠道噬菌体组中,以0.22μm滤膜为优。目前研究中发现,哺乳动物肠道中存在的Myoviridae与Siphoviridae等病毒,尺寸大于0.2μm,而最近发现的巨型真核病毒大小甚至约为400nm。相对的,如果为了保证病毒组活性,用于下游FVT(Faecal virome transplantation)等研究;或样本来源于复杂环境,为了避免遗漏巨型病毒信息,这样的研究建议以0.45μm滤膜为主。如Ling Deng 等在研究中为保证噬菌体活性而采用0.45μm滤膜。
Nádia Conceição-Neto等通过8种病毒及4种细菌对微生态群落进行模拟(表2),并采用不同的过滤,离心等条件,对不同的颗粒富集进行评估。在过滤条件的评估中,采用0.8μm离心滤膜(centrifugal filter, PES),0.8μm的聚碳酸酯滤膜(polycarbonate filter, PC),0.45μm的离心滤膜(centrifugal filter, PVDF)或0.22μm的离心滤膜(centrifugal filter, PVDF),并与未过滤的对照进行比较(图1)。结果表明,两种0.8μm和0.45μm滤膜对模拟病毒组中的大多数病毒呈现较小的影响。但对于mimivirus(模拟病毒组中最大的病毒),0.8μm PES 损失了81.7%,0.8μm PC损失 95.9%、0.45μm PVDF损失99.0%。然而使用0.22μm滤膜时,99.90%的mimivirus,与82.0%的herpesvirus出现了损失。相比病毒组,0.8μm PES、0.45μm 和0.22μm的滤膜皆有效地移除了99.5%到99.9%的细菌。
除了孔径之外,滤膜的材料还极大地影响过滤效率。在0.8μm的PC和PES滤膜之间可以明显观察到这一点。0.8μm的PC的孔精确地呈圆柱形,并且狭窄地分布在聚碳酸酯薄板上,而0.8μm PES滤膜是3D聚合物网络,因此对细菌和mimicvirus的过滤效率更高。

表2:常见病毒的大小(Nádia Conceição-Neto, et al, 2019)

图1: 不同过滤条件对病毒(A)及细菌(B)形成的影响(Nádia Conceição-Neto, et al, 2019)
氯仿作为常用的病毒颗粒纯化的化学试剂,具有破坏脂质膜稳定性,破坏细菌结构完整性的特征。 在上面提到的研究中(Nádia Conceição-Neto, et al, 2019),氯仿浓度与细菌去除效率高度相关。 但是,包膜病毒,特别是coronavirus 和mimivirus,在去除包膜后也失去了稳定性。 另外,rotavirus和polyomavirus等一些非包膜病毒也易受氯仿的影响。 众所周知,其他属于Corticoviridae,Plasmaviridae或Inoviridae的非包膜噬菌体同样对氯仿敏感。 氯仿的作用似乎具有高度的病毒特异性,因此用氯仿纯化病毒颗粒可能不是病毒学研究的首选方法。
不同方法对病毒颗粒富集的影响
除了滤膜之外,用化学试剂来去除宿主细胞和细菌等污染,也是病毒颗粒富集中常用的方法,如通过CSCl梯度密度离心等Manuel Kleiner等在Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes文章中通过在无菌小鼠中植入六种病毒噬菌体:P22、T3、T7、ɸVPE25、ɸ6和M13,其中P22、T3、T7、 ɸVPE25代表四种dsDNA病毒,M13代表ssDNA病毒,ɸ6代表ssRNA病毒 ;两种细菌:革兰氏阳性菌 Listeria monocytogenes EGD-e和革兰氏阴性菌Bacteroides thetaiotaomicron VPI5482来模拟肠道微生态环境。其中六种噬病毒菌体为等数量加入,两种细菌为等数量加入;最后总噬菌体数量和总细菌数量相等。之后对以下五种VLPs富集方法进行了评估,包括:(i)FD: 0.45μm滤膜过滤去除微生物细胞+DNase消化去除游离DNA; (ii)DTT: 二硫苏糖醇(Dithiothreitol)处理以降解粪便粘液+过滤+Dnase; (iii)PEG: 过滤+DNase+聚乙二醇(polyethylene glycol)进行噬菌体颗粒浓缩沉淀; (iv)CsCl: 过滤+ DNase + CsCl密度梯度离心以纯化噬菌体; (v)MG: 宏基因组提取方法。其中由于PEG 加入滤液时形成了粘稠的高分子化合物,而这种化合物在后续操作中无法去除而导致PEG实验方法失败。从而结果只对其中四种方法进行了比较(图2)。

图2:VLPs纯化方法示意图(Kleiner M, et al, 2015)
从结果中可以看出(图3 ),在未经富集的VLPs样本得到的测序数据中,比对到噬菌体病毒的reads只占不到5%,而VLPs经三种富集方式(FD, DTT 和CsCl)处理的样本得到的数据中比对到噬菌体的reads均大于80%,并且用CsCl方法富集的样本宿主去除率最高,其次是DTT富集方法,最后是FD方法。
将测序数据中比对到小鼠宿主的数据滤掉之后重新计算各自的数据占比,如图3B所示,用CsCl方法富集的样本数据中比对到噬菌体P22的reads数明显偏高,比对到ɸVPE25的reads则偏少,说明通过CsCl梯度离心方式富集粪便样本时,对不同密度的噬菌体有较为明显的偏好性。

图3:不同方法下人工肠道微生态中相对reads丰度(A),和基于基因组大小校正后的相对reads丰度(B)(Kleiner M, et al, 2015)
结合表3可以看出,与未经病毒类似物富集的实验方法比,三种纯化方式在去除宿主DNA和细菌DNA均是非常成功的,富集倍数均可达到40000倍以上。其中CsCl方法的富集效果最佳,达到49077倍,FD富集效果最弱,但也有41517倍。这些变化的富集倍数均可表明,在去除细菌DNA时,这三种纯化方式的去除率均可达到99.99%以上。
在粪便组织样本的研究中,宿主DNA的污染对VLPs研究的干扰也不容忽视,所以在选择纯化方式时,应将宿主DNA的去除效率也考虑进去。文章中采用的三种纯化方式,在宿主DNA的去除中差异较为明显。用比对到噬菌体的reads数与比对到小鼠的reads相比,FD只有55倍的变化,意味着该方法对宿主DNA的去除率为98.1%,而CsCl有768的富集倍数,表示去除率为99.87%。
表3:不同纯化方法对小鼠和细菌DNA的去除效率(Kleiner M, et al, 2015)
文章中,作者主要关注的是dsDNA噬菌体基因组的测序结果,然而在这个人工模拟肠道微生物群体中,作者也加入了两种非双链DNA病毒 ɸ6和M13用来验证dsRNA病毒和ssDNA病毒的基因组是否可以用以上测试方法获得。为了确定测序结果中ɸ6噬菌体序列的测序情况,作者把FD和MG方法获取的样本分别进行了反转录并合成cDNA。但是由于样本中ɸ6噬菌体的RNA含量过低而导致建库失败,所以在包含MG方法在内的所有纯化方式获得测序结果中,均没有测到属于ɸ6噬菌体的reads。但是,在所有的测试样本中均测到了一小部分ssDNA病毒M13的序列。理论上,按照Illumina文库制备过程,只有双链DNA才可以进入建库流程,单链DNA由于不能连上测序接头,因而不能进入测序阶段的。通常来讲,建库时用于连接Illumina测序接头的T4 DNA连接酶只对dsDNA起作用,但从测序比对结果可以看出,T4 DNA ligase也可以作用于ssDNA,尽管此时的连接效率非常低,这可能用来解释为什么测到了部分ssDNA病毒的序列。这些结果也表明,如果有足够多的reads产生,上述几种纯化方式都可以用于定性的评估ssDNA病毒的存在情况。但是若要更加清晰的了解ssDNA和RNA病毒的存在状态,还需要采取其他研究手段。
在该研究中,作者利用已知细菌和病毒组成的人工肠道微生物样本,对VLPs的四种纯化方式进行了评估。除了PEG法在纯化过程中失败了,其他三种方法在不同方面各有优势。与FD和DTT法相比,CsCl纯化方式在去除宿主DNA时有明显的优势,但该方法对特殊噬菌体的选择偏好性较为明显,而FD和DTT在纯化时表现出了更好的均一性,并且DNA获得率也更高;FD和DTT两种方法获得结果差异较小,并且当粪便样本滤液较为粘稠时可采取DTT纯化方法,以降低滤液粘稠度,减少滤膜阻塞。虽然PEG在此研究中并无评估,但其对蛋白杂质的去除作用,也有研究证明,PEG处理对荚膜类病毒造成一定损失。另外在设计病毒宏基因组的研究实验时,还有一个至关重要的因素需要考虑,就是在实验时需要加入阴性对照以去除纯化、建库和测序时引入的污染干扰。
在另外一个研究中,Dennis S. Nielsen课题组对噬菌体颗粒富集的方法进行了优化。研究中对切入流过滤(tangential-flow filtration, TFF)和聚乙二醇沉淀(PEG)两种从粪便样品中提取噬菌体的方法进行了优化,并与之前发表的方法进行了比较(literature-adapted protocol, LIT)。为了量化噬菌体回收率,在提取过程的每个步骤中,都向样品中掺入少量的c2,φ29和T4噬菌体(分别为Siphoviridae,Podoviridae和Myoviridae科的代表)并观察噬斑形成单位(plaque forming unit, PFU)。与LIT相比,TFF和PEG对所有加入的噬菌体的回收率更高,每体积产生的噬菌体颗粒最多增加16倍,噬菌体DNA最多增加68倍,从而增加了提取低丰度噬菌体的机会。TFF和PEG衍生的宏病毒组数据显示,感染肠道相关细菌的Caudovirales和未分类噬菌体的相对丰度增加了10%(TFF和PEG大于92%,LIT大于82.4%)。与LIT(22%)相比,TFF与PEG的方法获得了较低的Myoviridae科的相对丰度(<16%)。但这并不是由噬菌体本身的损失所造成的比例降低,而是由于Siphoviridae噬菌体的提取效率的增高所导致的(TFF和PEG> 32.5%,LIT为22.6%)。最后,该研究通过透射电子显微镜验证了通过TFF和PEG提取的样品中,噬菌体多样性显著提高(图4)。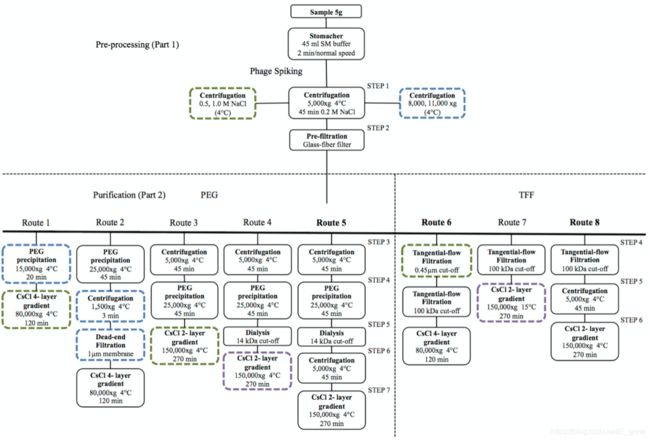
图4优化噬菌体提取方案的策略 (Josué L, et al, 2015)。
从图4可以了解,在VLPs富集中,预处理部分用于去除和沉淀大颗粒;纯化部分旨在去除低分子量杂质和微生物细胞。用虚线画出的方框表示不适合提取噬菌体的步骤(主要由于加入的噬菌体大量损失)。绿线框表示通过目测或TEM观察手,样本纯度极低,特别是低分子量杂质含量高。蓝线框表示有≥50%的加入的噬菌体数量丢失。紫色线框表示存在微生物污染。纯化程序的优化始于途径1(用于PEG方法)和途径6(用于TFF方法)。在整个优化过程中,将策略不断调整,直到加入的噬菌体的回收效率最高。策略6表示对照方法。最后根据回收效率,策略5(PEG)和策略8(TFF)成为了最优的富集方法。由此可见,VLPs富集过程中,离心速率,化学试剂,透析等方法的选择,都会影响病毒组的富集效率。
宏转录组与病毒颗粒富集方法的比较
研究病毒宏基因组学的最广泛使用的方法依赖于VLPs富集和测序前的基因组扩增。对于检测低基因比例的病毒,通常需要去除非病毒宿主基因组和细菌核酸。最近,通过对环境样品的总非核糖体RNA的测序,使得病毒的宏转录组成功地用于阐明各种无脊椎动物和脊椎动物的病毒组特征。Rowena Chong et al通过对野生和圈养的袋獾粪便分别进行VLPs富集后宏基因组测序和粪便的宏转录组测序,在阐明不同环境对粪便病毒组造成的影响的同时,也对两种方法进行了比较(Chong R, et al, 2020)。
总体而言,VLPs富集后宏基因组和宏转录组的结果无论是在检测到的病毒,或通过RSEM分析测得的转录本丰度皆有所不同(图5c和d)。宏转录组方法检测到的病毒组范围更广,其中49.87%到97.51%的病毒与RNA病毒最接近,而2.49%到50.13%的病毒与DNA病毒最接近。相反,VLPs富集后宏基因组的结果中,有95.54%的病毒相关序列与DNA病毒的最为接近,只有5%的病毒被确定为RNA病毒(图5b)。宏转录组方法揭示了高水平的病毒多样性,其中检测到的最丰富的病毒组是Caudovirales,Luteo-Sobemo,Narna-Levi,Partiti-Picobirna,Picorna-Calici和Tombus-Noda(图5c)。相反,VLPs富集后宏基因组方法揭示了相对较低的病毒多样性。Caudovirales占病毒reads中的主要比例(69.89%至99.49%),而Microviridae, Circoviridae, Genomoviridae, Parvoviridae, Herpesviridae, Polyomaviridae, 和Papillomaviridae检测到的丰度较低(图5c)。
通过宏转录组方法检测到的脊椎动物病毒占总病毒reads的0至9.41%。而其他大部分病毒reads属于非脊椎动物的真核病毒(45.08%至97.51%),其中包括植物病毒,昆虫病毒和霉菌病毒,或来自于Siphoviridae, Podoviridae, Myoviridae, 和Microviridae科的噬菌体(2.48%至48.91%)。VLPs富集后宏基因组的数据结果中,脊椎动物病毒reads的百分比也很小(0.04至0.84%),而噬菌体和其他真核病毒的读数分别在79.17%至99.91%和0.04%至19.99%之间。
通常,VLPs富集后宏基因组方法主要检测DNA病毒,而宏转录组方法检测DNA和RNA病毒,虽然器检测到的DNA病毒仅限于丰度相对较高的病毒(图5d)。高丰度水平通常表明病毒感染活跃,在此期间,DNA病毒被转录为可通过RNA测序检测到的RNA中间体。相反,即使根据RSEM估算的counts非常丰富,通过VLPs富集后宏基因组方法,也无法检测到在宏转录组方法中鉴定出的RNA病毒。
VLPs富集后的基因组随机扩增虽然增加了每个文库中病毒reads的数量。但是,样本的数据结果显示,其病毒组成都高度偏向DNA病毒,特别是来自Caudovirales科的噬菌体。在上文中介绍的比较各种富集方法的研究中发现,噬菌体占所有测试富集方法中所有reads的80%,未富集的样本中噬菌体占5%。尽管基因组随机扩增可以显著增加病毒reads的总数,但随机扩增的方法还是容易让结果产生偏倚。由于某些病毒序列的优先扩增,导致检测到某些其他病毒更少,且基因组覆盖率较低。然而,不论其已知的偏倚如何,VLPs富集后宏基因组学方法在研究病毒组表征中具有优势,因为它具有识别低丰度DNA病毒的能力,这尤其适用于检测休眠或无活性的病毒。
VLPs富集后宏基因组学方法相反,宏转录组方法是非病毒特异性的,可揭示样品中的整个转录组信息。由于没有VLPs富集且样品处理过程相对简单,因此用宏转录组学方法研究病毒组,有可能降低检测偏倚的可能性。在Rowena Chong等的这项研究中,通过宏转录组学测序的病毒reads的比例平均占每个文库的2%,但去除宿主后,可检测到的病毒组的种类和数量,显着高于VLPs富集后宏基因组学方法检测到的RNA和DNA病毒。重要的是,该项研究的结果结果表明,两种方法所揭示的病毒群落的分类组成不可互换,而且都无法检测到所有存在的病毒。因此,两种方法同时使用,是获得样本中病毒群落分类和功能概况的有力工具(Chong R, et al, 2020)。 
图5:VLPs富集后宏基因组学与宏转录组学方法下袋獾粪便病毒组特征。图a、b、c中上部分为宏转录结果,下部分为VLPs富集后宏基因组学结果(Chong, et al, 2019)
三代测序在宏病毒组中的应用
牛津纳米孔技术(Oxford Nanopore Technology , ONT)作为新兴的单分子实时测序技术(Single molecule real-time sequencing technology, SMRT)之一,具有快速制备文库,超长读取和实时数据采集等优势。对于病毒组,ONT测序有潜力通过产生覆盖单个病毒颗粒内所有突变的基因组长度读数来获得病毒基因组。此外,纳米孔测序不仅能够区分基因组,而且还能区分单碱基修饰,例如胞嘧啶的5-甲基化(DNA为5mC,RNA为m5C)和腺嘌呤的6-甲基化(DNA为6mA,RNA为m6A)。近年来,越来越多的证据表明DNA/RNA甲基化可以影响生物学功能,包括调节DNA/RNA复制和修复以及基因表达。最近,Oliveira 等报道艰难梭菌中的DNA甲基转移酶能够介导孢子形成,艰难梭菌疾病的传播和致病性(Oliveira, et al, 2020)。Xue等报道了病毒m6A可以上调人类呼吸道合胞病毒的复制和发病机制。这些发现表明表观遗传调控对于重要病原体的发病机制非常重要(Xue MG, et al, 2019)。
为了解析健康成年人的肠道病毒组,包括物种组成和潜在的表观遗传信息,中国科学院微生物研究所王军教授课题组开发了一种新的工作流程(Jiabao Cao, et al, 2020),该流程包括病毒粒子的物理富集,核酸的逆转录和扩增以及生物信息学分析,并首次使用ONT PromethION平台对五个健康的人类个体进行病毒组的表征(图6)。
结果表明,使用Nanopore测序病毒DNA和扩增后的病毒DNA/cDNA可获得更长读长的序列,提供有关病毒多样性的更多信息,且许多病毒序列并不在当前数据库中。此外,使用Nanopore测序非扩增的病毒DNA可以对噬菌体的表观遗传修饰(甲基化)信号进行检测。相较于传统的二代测序,ONT可以进行测序并几乎同时产生reads,可以用五天的工作时间完成从样品中生成数据,如果PromethION运行时间较短,或者在生成过程中进行实时数据分析,则可能更短。
然而,要利用ONT测序的能力,需要非常高浓度的DNA或RNA文库来产生足够的reads,这对于病毒DNA/RNA非常困难,因为在1.5g粪便样本中病毒DNA/RNA的含量通常只能达到输入量的10%。虽然基因组随机扩增会提高文库的输入量,并能够检测出低丰度的病毒,但由于随机引物的偏好性以及PCR产生的假阳性,导致产生了平均较短的reads并且对病毒丰度评估也存在偏差。最后,扩增的cDNA / DNA丢失了病毒核苷酸上的所有甲基化修饰,阻碍了表观遗传信息的研究,也大大降低了ONT测序平台的优势。因此,研究者需要平衡扩增过程的利弊,可以同时使用原始DNA(和/或RNA)和扩增的cDNA / DNA进行测序,从而在同一测序过程中获得互补信息。
此研究第一次通过直接测序证明噬菌体基因组存在甲基化。已知RSV病毒和流感病毒在其RNA基因组中具有m6A甲基化,此研究中的结果发现,噬菌体也以6mA作为DNA甲基化的主要形式。由于DNA甲基化在细菌对噬菌体的防御中起着重要作用,因此,噬菌体基因组如何甲基化,其对噬菌体生命周期的影响以及与细菌宿主的相互作用仍有待于专门研究中进行探讨
。
图6:病毒contig中不同甲基化位点识别和motif识别。(A)contig00000015中甲基化位点的分布(5mC和6mA); (B)5mC motif; ©6mA motif (Jiabao Cao, et al, 2020)
Reference:
Altschul SF, Gish W, Miller W, et al. Basic Local Alignment Search Tool[J]. Journal of Molecular Biology, 1990, 215(3): 403-410.
Amann RI, Ludwig W, Schleifer KH. Phylogentic Identification and In-situ Detection of Individual Microbial-cells Without Cultivation[J]. Microbiological Reviews, 1959, 59(1): 143-169.
Angly Florent E, Felts Ben, Breitbart Mya, et al. The marine viromes of four oceanic regions[J]. Plos Biology, 2006, 4(11): 2121-2131.
Breitbart M, Hewson I, Felts B. Metagenomic analyses of an uncultured viral community from human feces[J]. Journal of Bacteriology, 2003, 185(20): 6220-6223.
Casjens, S. Prophages and bacterial genomics: what have we learned so far?[J]. Molecular Microbiology, 2003, 49(2): 277-300.
Chong R, Shi M, Grueber CE, et al. Fecal Viral Diversity of Captive and Wild Tasmanian Devils Characterized Using Virion-Enriched Metagenomics and Metatranscriptomics[J]. Journal of Virology, 2019, 93(11).
Conceicao-Neto, Nadia; Zeller, Mark. Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis[J]. SCIENTIFIC REPORTS,2015, 5: 2045-2322
Flewett TH, Bryden AS, Davies H. Diagnostic Electron-Microscopy of Feces. 1. Viral Flora of Feces as Seen by Electron-Microscopy[J]. Journal of Clinical Pathology, 1974,27(8): 603-614.
Forsberg, Kevin J, Reyes Alejandro, Wang Bin. The Shared Antibiotic Resistome of Soil Bacteria and Human Pathogens[J]. Science, 2011, 337(6098): 1107-1111.
Gregory Ann C, Zayed Ahmed A, Conceicao-Neto Nadia et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole[J]. Cell, 2019, 177(5), 1109-+.
Jiabao Cao,Yuqing Zhang,Min Dai,Jiayue Xu,Liang Chen,Faming Zhang,Na Zhao,Jun Wang. Profiling of Human Gut Virome with Oxford Nanopore Technology[J]. Medicine in Microecology, 2020,4.
Josué L. Castro-Mejía, Musemma K. Muhammed, Witold Kot, et al. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome, 2015, 3(1).
Kazankov K, Barrera F, Moller HJ. Soluble CD163, a Macrophage Activation Marker, Is Independently Associated With Fibrosis in Patients With Chronic Viral Hepatitis B and C[J]. 2014, 60(2): 521-530.
Kim Joungmok, Kundu Mondira, Viollet Benoit. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1[J]. Nature Cell Biology, 2011, 13(2): 132-U71.
Kleiner M, Hooper LV, Duerkop BA. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes[J]. BMC Genomics, 2015, 7: 1471-2164
Letarov A, Kulikov E. The bacteriophages in human-and animal body-associated microbial communities[J]. Journal of Applied Microbiology, 2009, 107(1): 1-13.
Mazmanian SK, Round JL, Kasper, DL. A microbial symbiosis factor prevents intestinal inflammatory disease[J]. Nature, 453(7195): 620-625.
Minot Samuel, Sinha Rohini, Chen Jun. The human gut virome: Inter-individual variation and dynamic response to diet[J]. Genome Research, 2011, 21(10): 1615-1625.
Oliveira PH, Ribis JW, Garrett, EM. Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis[J]. Nature Microbiology, 2020, 5(1): 166-+.
Qin JJ, Li RQ, Raes, J,et al. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature, 2010, 464(7285): 59-U70.
Rajilic-Stojanovic M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota[J]. FEMS Microbiology Reviews, 2014, 38(5): 996-1047.
Rappe MS, Giovannoni SJ. The uncultured microbial majority[J]. Annual Review of Microbiology, 2003, 57: 369-394.
Rohwer Forest, Thurber Rebecca Vega. Viruses manipulate the marine environment[J]. Nature, 2009, 459(7244): 207-212.
Rowena Chong,Catherine E. Grueber,Samantha Fox,Phil Wise,Vanessa R. Barrs,Carolyn J. Hogg,Katherine Belov. Looking like the locals - gut microbiome changes post-release in an endangered species[J]. Animal Microbiome, 2019, 1(1).
Wexler HM. Bacteroides: the good, the bad, and the nitty-gritty[J]. Clinical Microbiology Reviews, 2007, 20(4): 593-+.
Xue, MG; Zhao, BS; Zhang, ZJ; Lu, MJ; Harder, O; Viral N-6-methyladenosine upregulates replication and pathogenesis of human respiratory syncytial virus[J]. Nature Communications. 2019. 10: 2041-1723.

