单细胞分析:归一化和回归(八)
导读
现在有了高质量的细胞,首先探索数据并确定任何不需要的变异来源。然后需要对数据进行归一化,计算方差并回归任何对数据有影响的协变量。
1. 学习目标
- 学会如何执行归一化,方差估计,鉴定易变基因
2.Info
- 目标
- 准确归一化和缩放基因表达值,以解决测序深度和过度分散计数值的差异。
- 识别最可能指示存在的不同细胞类型的变异基因。
- 挑战
检查并删除不需要的变异,这样就不用在下游对这些细胞进行聚类
- 建议
- 在执行聚类之前,对存在的细胞类型的期望有一个很好的了解。了解是否期望细胞类型复杂性较低或线粒体含量较高,以及细胞是否正在分化。
- 如果需要并且适合实验,则回归
UMI的数量(默认使用sctransform)、线粒体含量和细胞周期,因此不要驱动下游的聚类。
3. Set-up
首先为规范化和集成步骤,创建一个新脚本(文件 -> 新文件 -> R 脚本),并将其保存为SCT_integration_analysis.R。
对于工作流程的其余部分,将主要使用Seurat包中提供的功能。因此,除了tidyverse库和下面列出的其他一些库之外,还需要加载Seurat库。
# Single-cell RNA-seq - normalization
# Load libraries
library(Seurat)
library(tidyverse)
library(RCurl)
library(cowplot)
此分析的输入是seurat对象。将使用在QC课程中创建的filters_seurat 。
4. 变异溯源
对生物协变量的校正用于挑选出特定感兴趣的生物信号,而对技术协变量的校正可能对于揭示潜在的生物信号至关重要。最常见的生物学数据校正是去除细胞周期对转录组的影响。这种数据校正可以通过针对细胞周期分数的简单线性回归来执行。
第一步是探索数据,看看是否观察到数据中的任何影响。细胞之间的原始计数不具有可比性,不能直接使用它们进行分析。因此,将通过除以每个细胞的总计数并取自然对数来执行粗略的标准化。这种标准化仅用于探索当前数据中变异的来源。
注意:
Seurat最近引入了一种名为sctransform的归一化方法,该方法同时执行方差稳定并消除不需要的变化。这是目前工作流程中实施的方法。
# 归一化
seurat_phase <- NormalizeData(filtered_seurat)
接下来,获取这些标准化数据并检查是否需要数据校正方法。
5. 影响评估
要根据每个细胞的 G2/M 和 S 期标记的表达为每个细胞分配一个分数,可以使用Seuart函数CellCycleScoring()。此函数根据输入的canonical markers计算细胞周期阶段分数。
在 data文件夹中为您提供了一个人类细胞周期标记物列表,作为Rdata文件,称为cycle.rda。但是,如果您不使用人类数据,还有其他材料[1]详细说明如何获取其他感兴趣的生物的细胞周期标记。
# 加载细胞周期 markers
load("data/cycle.rda")
# 为细胞进行细胞周期评分
seurat_phase <- CellCycleScoring(seurat_phase,
g2m.features = g2m_genes,
s.features = s_genes)
# 查看分配给细胞的细胞周期分数和阶段
View([email protected])
在对细胞进行细胞周期评分后,使用PCA确定细胞周期是否是数据集中变异的主要来源。要执行PCA,需要首先选择特异性的特征,然后对数据进行缩放。由于高表达的基因表现出特异性,并且不希望“特异性基因”仅反映高表达,因此需要缩放数据以缩放随表达水平的变化。Seurat ScaleData()函数将通过以下方式缩放数据:
- 调整每个基因的表达,使细胞间的平均表达为 0
- 缩放每个基因的表达以使跨细胞的方差为 1
# 鉴定特异基因
seurat_phase <- FindVariableFeatures(seurat_phase,
selection.method = "vst",
nfeatures = 2000,
verbose = FALSE)
# 缩放表达
seurat_phase <- ScaleData(seurat_phase)
注意:对于
selection.method和nfeatures参数,指定的值是默认设置。
现在,可以执行PCA分析并将前两个主成分相互绘制。按细胞周期阶段划分数字,以评估相似性或差异。
# 执行 PCA
seurat_phase <- RunPCA(seurat_phase)
# 可视化
DimPlot(seurat_phase,
reduction = "pca",
group.by= "Phase",
split.by = "Phase")

基于这个图,没有看到很大的差异,不会回归由于细胞周期引起的变化。
6. SCTransform
使用SCTransform归一化和回归不需要的变异。
现在可以使用sctransform方法作为更准确的归一化方法,估计原始过滤数据的方差,并识别特异基因。sctransform方法使用正则化负二项式模型对UMI计数进行建模,以消除由于测序深度(每个细胞的总 nUMI)引起的变化,同时根据具有相似丰度的基因的汇集信息调整方差(类似于一些Bulk RNA-seq的 方法)。
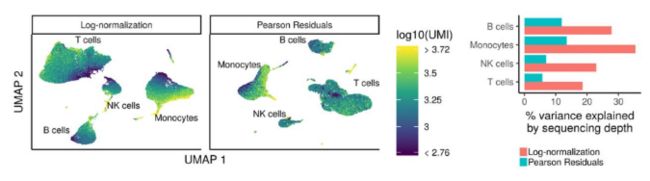
模型的输出(残差)是每个测试转录本的归一化表达水平。
Sctransform通过回归测序深度 (nUMIs) 自动计算细胞测序深度。但是,如果在探索步骤期间在数据中发现了其他无趣变化的来源,也可以包括这些来源。由于细胞周期阶段,观察到几乎没有影响,因此选择不从数据中回归。观察到线粒体表达的一些影响,因此选择从数据中回归。
为了运行 SCTransform,以下面的代码为例。
# SCTranform 不需要运行,仅展示
seurat_phase <- SCTransform(seurat_phase, vars.to.regress = c("mitoRatio"))
7. 迭代
- 迭代数据集中的样本
由于数据集中有两个样本(来自两个条件),希望将它们保持为单独的对象并转换它们,因为这是集成所需的。首先将seurat_phase对象中的单元格拆分为“Control”和“Stimulated”:
# 按条件拆分 seurat 对象以对所有样本执行细胞周期评分和 SCT
split_seurat <- SplitObject(seurat_phase, split.by = "sample")
split_seurat <- split_seurat[c("ctrl", "stim")]
现在将使用“for 循环”在每个样本上运行SCTransform(),并通过在SCTransform()函数的vars.to.regress 参数中指定来回归线粒体表达式。
在运行这个 for 循环之前,如果有一个大型数据集,那么可能需要使用以下代码调整 R 内允许的对象大小的限制(默认为 500 * 1024 ^ 2 = 500 Mb):
options(future.globals.maxSize = 4000 * 1024^2)
现在,运行以下循环来对所有样本执行sctransform。这可能需要一些时间(约 10 分钟):
for (i in 1:length(split_seurat)) {
split_seurat[[i]] <- SCTransform(split_seurat[[i]], vars.to.regress = c("mitoRatio"))
}

注意:默认情况下,在归一化、调整方差和回归无意义的变异来源之后,
SCTransform将按残差对基因进行排序,并输出 3000 个变异最多的基因。如果数据集具有较大的单元数,则使用variable.features.n参数将此参数调整得更高可能会有所帮助。
请注意,输出的最后一行指定“将默认检测设置为 SCT”。可以查看存储在seurat对象中的不同assays。
# # 检查哪些assays存储在对象中
split_seurat$ctrl@assays
现在可以看到,除了原始RNA计数之外,现在的检测槽中还有一个SCT组件。最具可变性的特征将是存储在SCT分析中的唯一基因。当进行scRNA-seq分析时,将选择最合适的方法用于分析中的不同步骤。
8. 保存结果
在完成之前,将此对象保存到data/文件夹。回到这个阶段可能需要一段时间,尤其是在处理大型数据集时,最好将对象保存为本地易于加载的文件。
saveRDS(split_seurat, "data/split_seurat.rds")
# 加载rds到环境中
split_seurat <- readRDS("data/split_seurat.rds")
参考资料
[1]附件材料: https://hbctraining.github.io/scRNA-seq_online/lessons/cell_cycle_scoring.html
本文由 mdnice 多平台发布