单细胞基因可视化之热图的根本改造2
我们前面已经说过了单细胞marker/或者其他任意基因的热图的简单修饰(单细胞基因可视化之热图改造修饰1),仅仅是基于颜色和DoHeatmap函数的参数修饰。那么要对热图进行根本的改造,关键点在于提取表达矩阵,然后对分组进行注释,就可以使用heatmap或者ComplexHeatmap来做图了。
首先计算marker基因,然后选择自己需要展示的基因。
library(Seurat)
library(stringr)
library(dplyr)
all.markers <- FindAllMarkers(scedata,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.75)
markers <- c("ACKR1",
"RAMP2",
"CLDN5",
"TM4SF1",
"PLVAP",
"IFI27",
"CLU",
"AQP1",
"GNG11",
"VWF",
"CLEC14A",
"PECAM1",
"SELE",
"COL3A1",
"LUM",
"COL1A1",
"COL1A2",
"DCN",
"SFRP2",
"C1S",
"COL6A2",
"CXCL13",
"CFD",
"CCL5",
"CCL4",
"CXCR4",
"PTPRC",
"LYZ",
"CD52",
"HCST",
"CD3D",
"NKG7",
"GZMA",
"CD2",
"S100A14",
"KRT16",
"LY6D",
"LGALS7B",
"SFN",
"FXYD3",
"DSP",
"CALML3",
"PITX1",
"ALDH3A1",
"DAPL1",
"TRIM29",
"PKP1",
"PKP3",
"DSC3",
"ACTA2",
"TAGLN",
"TPM2",
"MYH11",
"RERGL",
"RGS5",
"FRZB",
"LBH")
sc_marker <- all.markers[markers,]
然后提取表达矩阵,并对细胞进行注释。
# 提取原始表达矩阵
exp <- GetAssayData(scedata, slot = "counts")
exp <- log10(exp + 1)
head(scedata$celltype)
new_celltype <- sort(scedata$celltype)
head(new_celltype)
cs_data <- as.matrix(exp[sc_marker$gene, names(new_celltype)])
ac=data.frame(cluster=new_celltype)
rownames(ac)=colnames(cs_data)
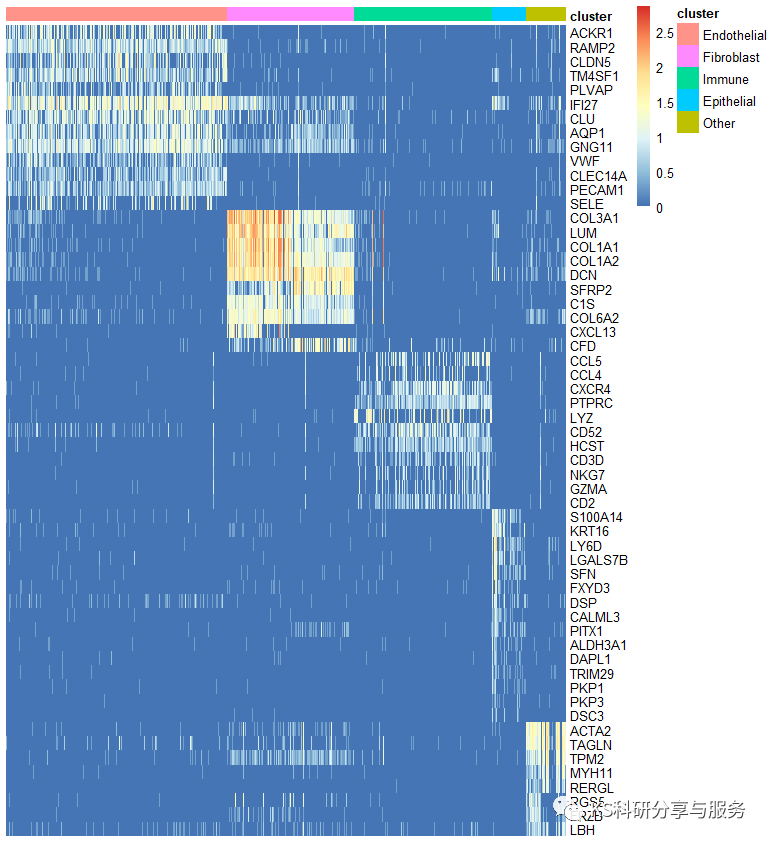
至此,我们需要作图的数据就已经得到了,可以使用热图函数进行绘制了。热图我们公众号之前讲的很详细了,可以参考,同样的做法。
library(pheatmap)
pheatmap(cs_data,show_colnames =F,show_rownames = T,
cluster_rows = F,
cluster_cols = F,
annotation_col=ac,
border_color = "black")
图片
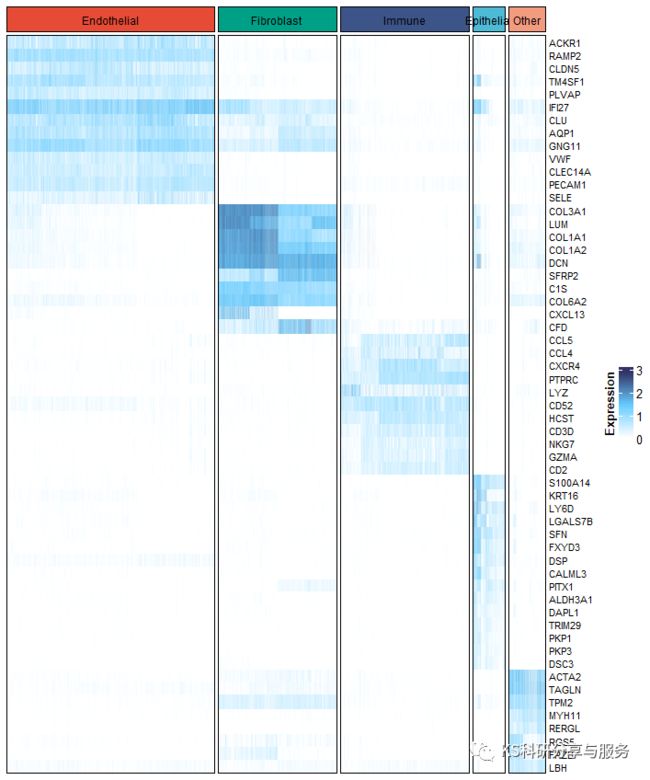
要进一步修饰,更加个性化的作图,采用ComplexHeatmap。
library(ComplexHeatmap)
color = paletteer_d("ggsci::nrc_npg")[c(1,3,4,2,5)]
names(color) <- levels(new_celltype)
top_anno <- HeatmapAnnotation(cluster=anno_block(gp=gpar(fill=color),
labels = levels(new_celltype),
labels_gp = gpar(cex=0.5,color='white',fontsize = 18)))
Heatmap(cs_data,
cluster_rows = FALSE,
cluster_columns = FALSE,
show_column_names = FALSE,
show_row_names = T,
column_split = new_celltype,
top_annotation = top_anno,
column_title = NULL,
heatmap_legend_param = list(
title='Expression',
title_position='leftcenter-rot'),
col = colorRampPalette(c("white","#66CCFF","#333366"))(100),
border = "black",
row_names_gp = gpar(fontsize = 8))
图片
看起来效果很好,心情舒适。今天分享就到这里了,整个热图也就这么些内容,想要其他更加个性化的修饰可以结合我们之前的内容自己调整。
觉得分享用的支持一下呗!不点赞、不分享还等什么!
更多内容请关注我的个人公众号《KS科研分享与服务》