《用于预测蛋白质-配体结合亲和力的结构感知交互图神经网络》
1.文章原标题与链接
《Structure-aware Interactive Graph Neural Networks for the Prediction of Protein-Ligand Binding Affinity》
原文链接
2.简介
药物发现通常依赖于蛋白质——配体结合亲和力的成功预测
图神经网络(GNNS)能实现更好的亲和预测
现有解决方案:**将蛋白质——配体复合物视为拓扑图数据
本文改进:结构感知的交互图形神经网络
1.偏光启动的图标注意(PGAL):迭代地志向节点边缘聚合过程以更新节点和边缘地嵌入
2.成对交互池(PIPOOL):收集交互式边缘
关键字:绑定关联预测;图神经网络;药物发现
3.引入
配体:通常是候选药物,包括小分子和生物制剂,它们可以在生物过程中与蛋白质相互作用,作为激动剂或抑制剂来治疗疾病
结合亲和力:定义为蛋白质和配体(如药物)之间的结合相互作用的强度,可以通过实验方法测量
生物测试是费力和耗时的。利用计算机辅助模拟方法和数据驱动学习模型,可以在药物发现的早期阶段预测绑定亲和力。结合亲和度的预测可以帮助对候选药物进行排序,并优先选择合适的药物进行后续测试,从而加快药物筛选的过程
三维结构信息可以有效地帮助药物设计
对接方法:在预测特定配体如何以可承受的计算成本与目标蛋白结合方面发挥着重要作用
优缺点:虽然对接过程能够较准确地识别蛋白-配体复合物的结合位形,但由于评分功能较差,其对结合亲和性的预测不准确、不可靠,这限制了对接方法在药物发现中的适用性
传统的机器学习方法:通过学习从蛋白质-配体复合物中提取的特征来提高性能。
优缺点:这些方法通用性有限,需要专家知识,并且严重依赖特征工程
基于深度学习的绑定亲和度预测:它将复杂数据表示为序列数据、三维网格数据或图数据,并利用各种神经网络进行预测。结构生物学中深度学习的关键挑战之一是如何建模三维空间结构以获得更好的性能。为此,现有的大多数工作都试图通过将复杂体作为3D网格表示来应用3D卷积神经网络(3D CNNs)。
优缺点:这些模型的成本是巨大的,尤其是考虑到远程交互时。此外,复杂结构中拓扑信息的缺失和对旋转的敏感性都对预测结果产生了负面影响。
使用gnn预测蛋白-配体结合亲和力的研究较少。许多研究者已经在药物发现的其他领域极大地发展了GNN模型,如预测分子性质和化学反应。现有药物设计中的gnn大多是通过结合距离信息来学习空间结构,这对于复杂体的三维结构建模是不够的。此外,目前的GNN框架无法处理蛋白质与配体之间的基础长程相互作用信息,这对预测结合亲和力有价值。
提出了一种新颖的结构感知交互式图神经网络(SIGN)来学习构建的复杂图。
两个设计组件:
1.用于建模3D空间结构的极向图形注意层(PGAL)
PGAL的核心思想是为每个中心目标建立一个极坐标系统,并在进行聚合过程时保持邻居的距离和角度信息。更具体地说,我们采用带图注意力的节点-边交互方案迭代集成空间因素,有效学习复杂体的三维结构。
2.用于利用远程交互的成对交互池(PiPool)
首先,由于蛋白质的体积较大,在复杂的图中包含完整的蛋白质结构是多余的,但这样也丢失了蛋白质与配体之间的长程交互信息。为了处理这个问题,SIGN的次要部分PiPool被设计成将这种全局交互合并到我们的模型中,它使用了一个感知类型的原子池进程在边缘引入辅助学习任务来重建原子相互作用矩阵
SIGN可以增强包含三维空间结构和全局交互作用的综合体的表示学习。
主要贡献:1.第一批从极坐标角度开发图神经网络用于基于结构的绑定亲和力预测的人
2.提出了一种新型的结构感知交互图神经网络(SIGN),该网络不仅可以通过极性启发图注意层(PGAL)捕获三维空间信息,还可以通过半监督方式通过两两交互池(PiPool)捕获全局远程交互
3.使用两个基准数据集进行了大量的实验,证明了我们的SIGN具有更好的泛化性
相关工作:首先回顾了预测蛋白配体结合亲和力的相关文献,然后详细介绍了图神经网络在药物发现中的最新进展。
4.模型框架(SIGN模型的整体框架与细节)
4.1综述:两大挑战:1.复杂图具有独特的结构 2.蛋白质和配体之间的远程相互作用对结合亲和力也至关重要
现有的GNN无法捕获这样的空间信息和交互
4.2极坐标启发的图形注意:为了模拟复杂体的三维结构,直观的方法是提供原子在GNN结构中的三维坐标。但是笛卡尔坐标系下的位置信息对平移和旋转都很敏感,导致模型在学习复杂表示时泛化能力较差。
我们采用迭代的节点→边缘和边缘→节点交互层,从空间分布的角度融合距离和角度信息
4.2.1极具启发的专注学习架构:极坐标由径长度r和极角组成.我们开发了一个基于交互的图注意网络,以利用协作框架中节点之间的距离和边之间的角度,如下图所示:
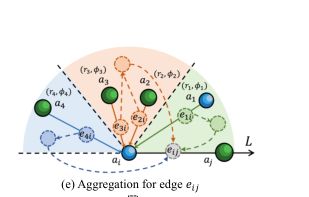
以eij为极轴进行划分,每个原子具有独一无二的极坐标,通过角域划分的方法,在边的第一个聚合阶段就可以通过角向注意来考虑复体的空间分布。
此外,距离因子也有助于结构建模,揭示空间相关性。原子之间距离的统计分布显示,共价键之间具有共价相互作用的长度主要集中在1-2A的范围内。聚合物中原子间的相互作用随距离的不同而不同,说明原子对的空间关系不同。我们可以建立原子间的距离图:
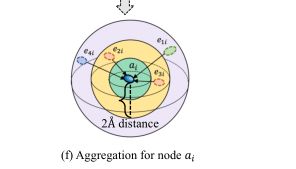
4.2.2角度导向 节点——>边交互层:在聚合过程中无法区分不同方向的邻居节点是现有GNN模型的一个缺点。为了克服这一不足,我们采用面向角度的图注意层来整合空间角度信息来更新边缘表示。
4.2.3距离感知 节点——>边交互层:将角度信息注入到边缘嵌入中后,将另一个空间因素纳入极坐标系统,即距离。
4.3成对交互池约束:由于图尺寸的限制和不必要的噪声,只包含部分蛋白质结构。然而,蛋白质与配体之间的长程分子间相互作用对结合亲和力有影响。为了捕捉复合物中的长程相互作用(例如碳碳共现相互作用),我们设计了一个感知原子类型的池层,用于蛋白质和配体之间的边缘,生成了原子类型对的邻近相互作用矩阵,并通过额外的自监督训练增强了表示学习过程。
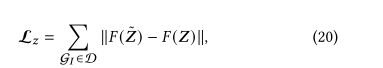
4.4优化结果:
将节点(原子)嵌入加在一起以得到复杂的表示,并使用MLP层作为回归因子来预测蛋白质与配体的结合亲和性
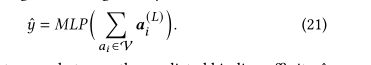
采用L1损耗函数对模型预测的结合亲和函数之间的绝对误差进行优化
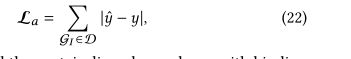
整合交互效果,更好地进行复杂表示学习,进一步结和复杂交互约束公式(20),得到总体目标函数如下:
![]()
5.实验
我们在两个标准数据集上进行实验,以调查以下研究问题:
1.与最新的模型相比,我们模型的性能如何
2.在大规模的低质量数据集训练时,泛化能力
3.空间和交互因素是否有利于预测
4.参数设置(如截止距离和角度域划分)如何影响预测结果?
5.1实验设置:
5.1.1数据集:
pdbbind是一个正在开发中的著名公共数据集,它提供了蛋白质配体配合物的三维结合结构,通过实验确定了结合亲和力(参见附录a .2)。在我们的实验中,我们主要使用的是PDBbind v2016数据集,该数据集是最近的工作中使用最频繁的。具体来说,它包括三个重叠子集,即一般子集、精细化子集和核心子集。一般集包含所有的13283个蛋白配体配合物,而精制集中的4057个配合物是从一般集中精选出来的质量更好的配合物。此外,通过仔细的选择过程,具有290个复合物的核心集作为测试的最高质量基准。为了方便起见,我们将细化的子集和核心子集(即3767个复合物)之间的区别称为下面pdbind的细化集。
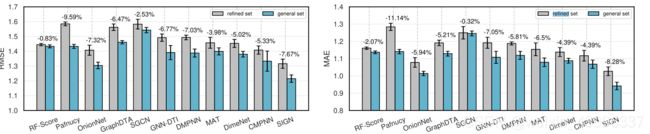
在常规集上进行培训时,pdbind基准测试的性能改进
MAE,平均绝对误差(Mean Absolute Error),观测值与真实值的误差绝对值的平均值。
RMSE:均方根误差(Root-mean-square error), 观测值与真值偏差的平方和与观测次数m比值的平方根。
CSAR-HIQ是一个额外的基准数据集,包含176和167蛋白配体复合物的两个子集。我们使用这个来自独立来源的外部数据集来进一步评估模型的泛化能力。
5.1.2设置:我们选择pdbind的精炼集作为我们的主要训练数据,因为完整的通用集和CSAR-HiQ数据集之间有相当多的重叠。我们以9:1的比例将蛋白质配体配合物随机分离,进行训练和验证。对于测试集,我们使用核心集和CSAR-HiQ集,去除精制集中存在的复合物。由于一般集质量较低的数据仍然可以提高模型的性能,我们在较大但质量较差的全一般集上进行了补充实验来分析我们的模型的泛化性。如上所述,由于CSAR-HiQ数据集存在重叠问题,我们只能在核心集上评估性能。接下来,我们从精炼集中随机选择1000个复合物作为验证集。其余的11,993个综合训练场用于训练。
5.1.3评价指标:使用均方根误差(RMSE)、平均绝对误差(MAE)、皮尔逊相关系数®和标准差(SD)来度量预测误差。
5.1.4比较:我们将我们提出的模型与比较方法进行比较,包括基于机器学习的方法(LR, SVR, RF-Score),基于cnn的方法(Pafnucy和onionnet),以及GNN模型GraphDTA用于蛋白质配体结合亲和力预测。此外,还比较了各种最先进的基于gnn的模型(SGCN,GNN-DTI,DMPNN,MAT, dimet和CMPNN),这些模型也考虑了分子建模的空间信息,以评价SIGN的性能。
5.2表现评估:
5.2.1:整体比较:我们首先在两个基准数据集上比较我们提出的SIGN与基线方法。如表2所示,在五次随机运行中报告了测试性能的四个指标的平均值和标准偏差。总的来说,我们可以观察到SIGN在两个数据集上取得了最好的性能,与pdbind和CSAR-HiQ数据集上的最佳基线模型相比,RMSE分别提高了6.5%和3.9%。
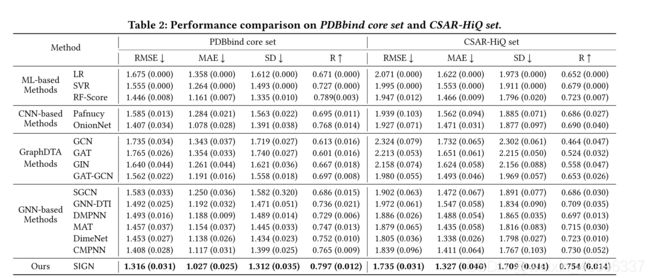
我们提出的SIGN不仅可以获取更全面的角度增强的结构信息,而不仅仅是距离,而且可以通过多任务学习框架处理复杂结构中的交互。因此,SIGN是一种非常有效的蛋白质-配体复合物建模方法,能够准确预测其结合亲和力。
5.2.2:通用性比较:
具有结合亲和力的基于三维结构的蛋白配体数据越来越多,而精细化集中高质量数据的数量相对较少。因此,利用更多质量较低的数据来提高绩效的能力表明了模型的通用性,这是绩效评价的另一个必要度量。如第5.1.2节所介绍的,我们对pdbind数据集的通用集进行了额外的泛化实验。如图5所示,我们将建议的SIGN与两个训练集上的主要竞争基线进行比较。结果表明,在两种训练条件下,SIGN的预测误差均显著最小。更重要的是,我们的模型在一般集上训练时提高了约8%的性能,并进一步扩大了与基线相比的预测优势。因此,对于数据量大但质量差的情况,SIGN具有更强的通用性。
5.2.3:空间及交互因素的影响:
为了验证影响最终性能的因素的有效性,我们在两个基准测试中比较SIGN及其变体
•SIGN-AD 使用没有角度和距离信息的GAT层进行节点边缘交互
•SIGN-Duses 使用没有距离信息的GAT层
•SIGN-A 使用无角度信息的GAT层
•SIGN-I 删除交互损失
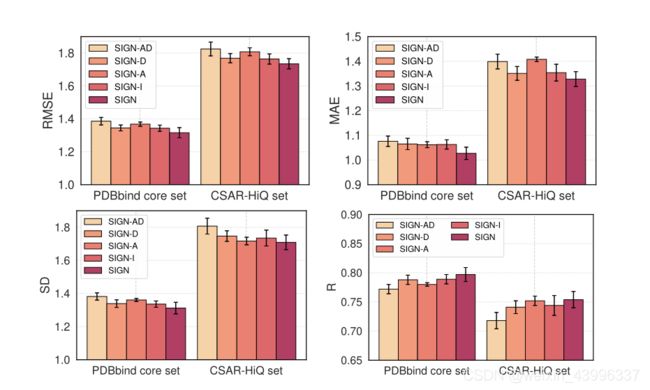
正如我们所看到的,所提出的SIGN优于其他变体,证明了协同处理空间和交互信息的必要性,这对蛋白质-配体结合亲和力预测至关重要
5.2.4:参数分析:
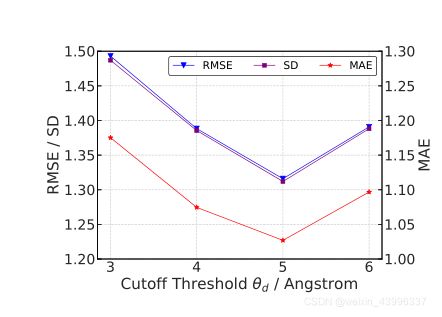
截止距离:
复杂图构造中截止距离在3-6倍波长范围内时,均方程误差讨论:截止距离在3-5倍波长范围内变化时,随着截止距离增大误差减小,这说明此范围内更多的空间信息可以用于我们的模型,并有利于更好地学习复杂表示,这导致性能显著改善。在此之后,过长的切断距离将引入额外的冗余和降低性能。
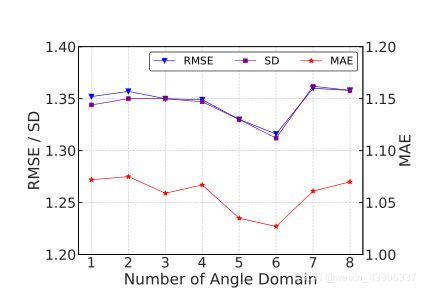
角域划分:
为了更深入地了解角度信息在我们的模型中的影响,我们将角度域从1划分到8。可以看出,当角域数为5或6时,模型性能最佳。太细粒度或太粗粒度的划分将导致性能下降。一种可能的解释是,太细粒度的划分无法提供空间中可识别的信息,而太大粒度的角度域包含非常稀疏的原子邻居,这两种情况都对空间信息的学习产生不利影响
六.结论
本文主要研究如何改进蛋白质与配体结合亲和力的预测。具体来说,我们提出了一个基于gnn的模型,SIGN,来学习蛋白质-配体复合物的表示,利用原子间的细粒度结构和相互作用信息,更好地预测结合亲和力。沿着这条线,我们设计了极向图注意层(PGAL)来整合距离和角度信息,用于三维空间结构建模。此外,为了进一步提高预测性能,我们引入了精心设计的池化过程和交互矩阵的重构学习任务。最后,在两个基准上的实验结果表明了该模型的有效性和通用性.