2018年SCI论文--整合GEO数据挖掘完整复现 六 :DAVID在线工具进行GO富集分析
文章目录
-
- 论文地址
- DAVID官网
- 上调基因GO富集分析
-
- 进入官网,点击“Function Annotation”选项
- 富集分析
- 选择背景基因
- 选择GO富集分析结果
- 下载富集分析结果
- 保存文件,作为后续可视化的输入文件
- 可视化富集分析结果
-
- bp,cc,mf分别提取counts数前5的term
- 条形图
- 圈圈图
- 下调基因GO富集分析
-
- 可视化富集分析结果
-
- bp,cc,mf分别提取counts数前5的term
- 条形图
- 圈图
论文地址
DAVID官网
上调基因GO富集分析
进入官网,点击“Function Annotation”选项

富集分析
1.“Enter Gene List”选项中,复制筛选过的111个上调基因的SYMBOL ID作为输入文件,
2.“Select Identifiter” 选项中,选择“OFFICIAL_GENE_SYMBOL”,作为输入基因ID名称
3.“List Type”选项中,选择“Gene List”
4.点击“Submit List”选项

选择背景基因
研究对象为人类癌症,所以选择“Homo sapiens”选项,点击“Use”,这时系统自动进行富集分析

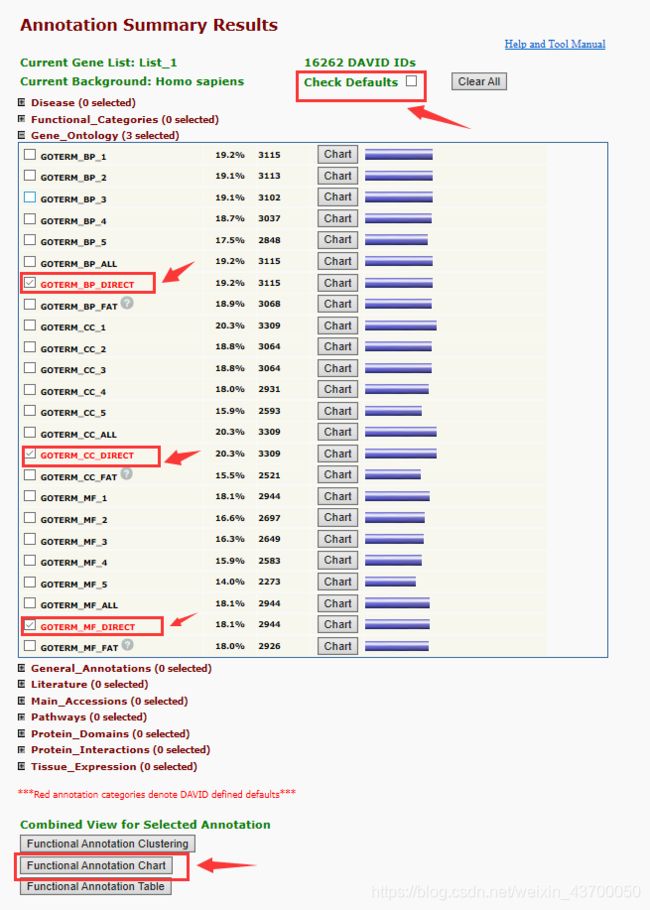
选择GO富集分析结果
1.首先取消“Check Defaults”选项,点击“Gene_Ontology(3 select)”下拉选项,
2.分别选择“GOTERM_BP_DIRECT”、“GOTERM_CC_DIRECT”、“GOTERM_MF_DIRECT”三个选项,
3.最后点击“Function Annotation Chart”选项,得到最终富集分析结果,
下载富集分析结果
点击“Download File”选项,


保存文件,作为后续可视化的输入文件
复制所有富集分析结果,保存为“up_GO.txt”文件

可视化富集分析结果
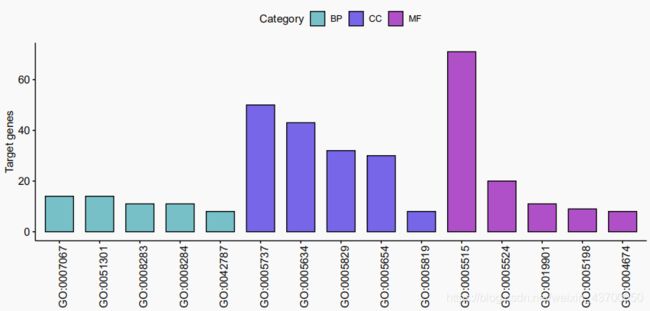
bp,cc,mf分别提取counts数前5的term
setwd("./3.DAVID_GO_KEGG/GO/UP_GO")
up = read.table(file = 'up_GO.txt',sep = '\t',header = T,quote = '')
up_rt = up[up$PValue < 0.05,]
library(tidyr)
up_rt = separate(up_rt, Term, sep = "~",
into = c("ID", "Term"))
bp_df = up_rt[up_rt$Category == 'GOTERM_BP_DIRECT',]
bp_df = bp_df[order(bp_df$Count,decreasing = T),]
bp = bp_df[1:5,]
cc_df = up_rt[up_rt$Category == 'GOTERM_CC_DIRECT',]
cc_df = cc_df[order(cc_df$Count,decreasing = T),]
cc = cc_df[1:5,]
mf_df = up_rt[up_rt$Category == 'GOTERM_MF_DIRECT',]
mf_df = mf_df[order(mf_df$Count,decreasing = T),]
mf = mf_df[1:5,]
allGo = rbind(bp,cc,mf)
library(stringr)
table(allGo$Category)
allGo$Category = substr(allGo$Category,8,9)
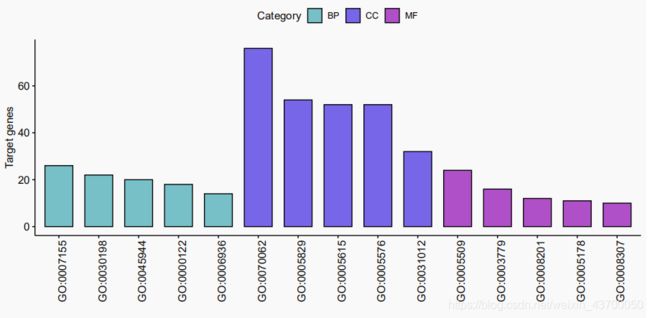
条形图
library(ggpubr)
colnames(allGo)
p = ggbarplot(data = allGo,x = "ID",y = 'Count',
fill = "Category",
palette = c("cadetblue3","mediumslateblue","mediumorchid3"),
sort.by.groups = T,xlab = '',ylab = "Target genes")
ggpar(p,x.text.angle = 90)
ggsave(plot = p,'barplot.pdf',width = 10,height = 5)
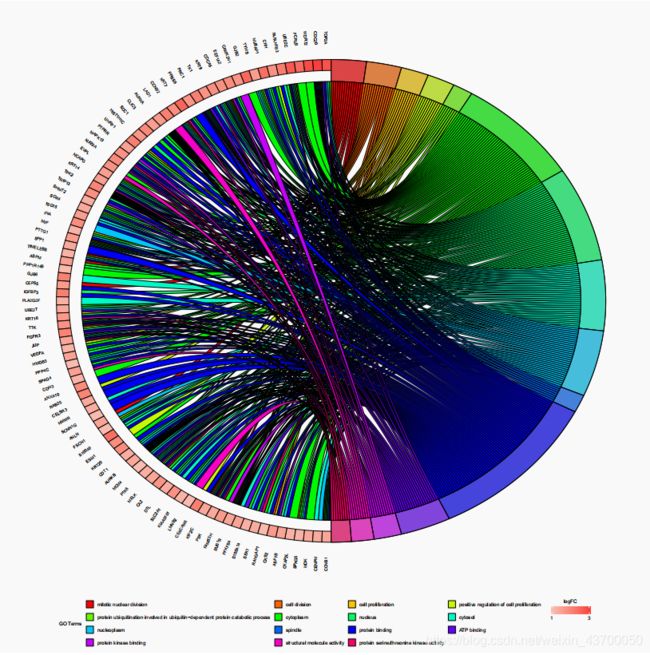
圈圈图
library(ggplot2)
library(GOplot)
upSig<-read.table("upSig.xls",sep="\t",header=TRUE,quote = '')
upSig = upSig[,c(1,4)]
colnames(upSig) = c('ID','logFC')
data = allGo[,c(1,2,3,7,6)]
colnames(data) = c('category', 'ID', 'term','genes','adj_pval')
circ <- circle_dat(data,upSig)
process<-data$term
chord <- chord_dat(circ, upSig,process)
up_circleplot = GOChord(chord)
ggsave(plot = up_circleplot,'up_circleplot.pdf',width = 20,height = 20)
下调基因GO富集分析
与上调基因富集分析步骤一样,得到“down_GO.txt”文件,在这里只显示可视化结果
可视化富集分析结果
bp,cc,mf分别提取counts数前5的term
setwd("./3.DAVID_GO_KEGG/GO/DOWN_GO")
down = read.table(file = 'down_GO.txt',sep = '\t',header = T,quote = '')
down_rt = down[down$PValue < 0.05,]
library(tidyr)
down_rt = separate(down_rt, Term, sep = "~",
into = c("ID", "Term"))
bp_df = down_rt[down_rt$Category == 'GOTERM_BP_DIRECT',]
bp_df = bp_df[order(bp_df$Count,decreasing = T),]
bp = bp_df[1:5,]
cc_df = down_rt[down_rt$Category == 'GOTERM_CC_DIRECT',]
cc_df = cc_df[order(cc_df$Count,decreasing = T),]
cc = cc_df[1:5,]
mf_df = down_rt[down_rt$Category == 'GOTERM_MF_DIRECT',]
mf_df = mf_df[order(mf_df$Count,decreasing = T),]
mf = mf_df[1:5,]
allGo = rbind(bp,cc,mf)
library(stringr)
table(allGo$Category)
allGo$Category = substr(allGo$Category,8,9)
条形图
library(ggpubr)
colnames(allGo)
p = ggbarplot(data = allGo,x = "ID",y = 'Count',
fill = "Category",
palette = c("cadetblue3","mediumslateblue","mediumorchid3"),
sort.by.groups = T,xlab = '',ylab = "Target genes")
ggpar(p,x.text.angle = 90)
ggsave(plot = p,'barplot.pdf',width = 10,height = 5)
圈图
library(ggplot2)
library(GOplot)
downSig<-read.table("downSig.xls",sep="\t",header=TRUE,quote = '')
downSig = downSig[,c(1,4)]
colnames(downSig) = c('ID','logFC')
data = allGo[,c(1,2,3,7,6)]
colnames(data) = c('category', 'ID', 'term','genes','adj_pval')
circ <- circle_dat(data,downSig)
process<-data$term
chord <- chord_dat(circ, downSig,process)
dowm_circleplot = GOChord(chord)
ggsave(plot = dowm_circleplot,'dowm_circleplot.pdf',width = 20,height = 20)

本博客内容将同步更新到个人微信公众号:生信玩家。欢迎大家关注~~~