GEO数据挖掘(二)
生信技能树学徒第二周
一、GEO数据库 芯片数据获取
#数据下载
rm(list = ls())
library(GEOquery)
#先去网页确定是否是表达芯片数据,不是的话不能用本流程。以GSE56649为例
gse_number = "GSE56649"
eSet <- getGEO(gse_number, destdir = '.', getGPL = F)
class(eSet)
length(eSet)
#得出往往是一个长度为一的列表,转换为数据框的格式
eSet = eSet[[1]]
#(1)提取表达矩阵exp
exp <- exprs(eSet)
dim(exp)
exp[1:4,1:4]
#检查矩阵是否正常,如果是空的就会报错,空的和有负值的、有异常值的矩阵需要处理原始数据。
#如果表达矩阵为空,大多数是转录组数据,不能用这个流程。
#自行判断是否需要log,一般boxplot 后的纵坐标范围在0-20之间,如果过高就需要取log
#exp = log2(exp+1)
boxplot(exp)
#(2)提取临床信息,会有分组信息,我们根据分组信息来进行命名
pd <- pData(eSet)
#(3)让exp列名与pd的行名顺序完全一致,
p = identical(rownames(pd),colnames(exp));p
if(!p) exp = exp[,match(rownames(pd),colnames(exp))]
#(4)提取芯片平台编号
gpl_number <- eSet@annotation;gpl_number
save(gse_number,pd,exp,gpl_number,file = "step1output.Rdata")因此,数据获取并确认的流程,getGEO函数下载数据→转换格式→提取表达矩阵→提取分组信息→确认分组对应
二、确定分组以及探针注释的获取
数据默认分组时冗长的,我们一般用control和treat来进行重新命名分组

# 标准流程代码是二分组
# 生成Group向量的三种常规方法,三选一,选谁就把第几个逻辑值写成T,另外两个为F。如果三种办法都不适用,可以继续往后写else if
if(F){
# 1.Group----
# 第一种方法,有现成的可以用来分组的列
Group = pd$`disease state:ch1`
}else if(F){
# 第二种方法,自己生成
Group = c(rep("RA",times=13),
rep("control",times=9))
Group = rep(c("RA","control"),times = c(13,9))
}else if(T){
# 第三种方法,使用字符串出理的函数获取分组
Group=ifelse(str_detect(pd$source_name_ch1,"control"),
"control",
"RA")
}
#结束之后及时进行检查
# 需要把Group转换成因子,并设置参考水平,指定levels,对照组在前,处理组在后
Group = factor(Group,levels = c("control","RA"))#确定control的位置,总是要把它放在第一的位置上!关键词相应
Group探针对应到基因里

#捷径,不同GPL 基因注释是不同的,因此获取相应的GPL编号
library(tinyarray)
find_anno(gpl_number)
ids <- AnnoProbe::idmap('GPL570')
#四种方法,方法1里找不到就从方法2找,以此类推。
#方法1 BioconductorR包(最常用)
gpl_number
#http://www.bio-info-trainee.com/1399.html
if(!require(hgu133plus2.db))BiocManager::install("hgu133plus2.db")
library(hgu133plus2.db)
ls("package:hgu133plus2.db")#展示这个包里有哪些数据
ids <- toTable(hgu133plus2SYMBOL)#专用函数获取数据
head(ids)
# 方法2 读取GPL网页的表格文件,按列取子集
##https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570
if(F){
#注:表格读取参数、文件列名不统一,活学活用,有的表格里没有symbol列,也有的GPL平台没有提供注释表格
b = read.table("GPL570-55999.txt",header = T,
quote = "\"",sep = "\t",check.names = F)
colnames(b)
ids2 = b[,c("ID","Gene Symbol")]
colnames(ids2) = c("probe_id","symbol")
ids2 = ids2[ids2$symbol!="" & !str_detect(ids2$symbol,"///"),]
}
# 方法3 官网下载注释文件并读取
##http://www.affymetrix.com/support/technical/byproduct.affx?product=hg-u133-plus
# 方法4 自主注释
#https://mp.weixin.qq.com/s/mrtjpN8yDKUdCSvSUuUwcA
save(exp,Group,ids,gse_number,file = "step2output.Rdata")三、PCA和热图绘制
rm(list = ls())
load(file = "step1output.Rdata")
load(file = "step2output.Rdata")
#输入数据:exp和Group
#Principal Component Analysis
#http://www.sthda.com/english/articles/31-principal-component-methods-in-r-practical-guide/112-pca-principal-component-analysis-essentials
# 1.PCA 图----
dat=as.data.frame(t(exp))
library(FactoMineR)
library(factoextra)
dat.pca <- PCA(dat, graph = FALSE)
pca_plot <- fviz_pca_ind(dat.pca,
geom.ind = "point", # show points only (nbut not "text")
col.ind = Group, # color by groups
palette = c("#00AFBB", "#E7B800"),#规定颜色
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups"
)
pca_plot
save(pca_plot,file = "pca_plot.Rdata")
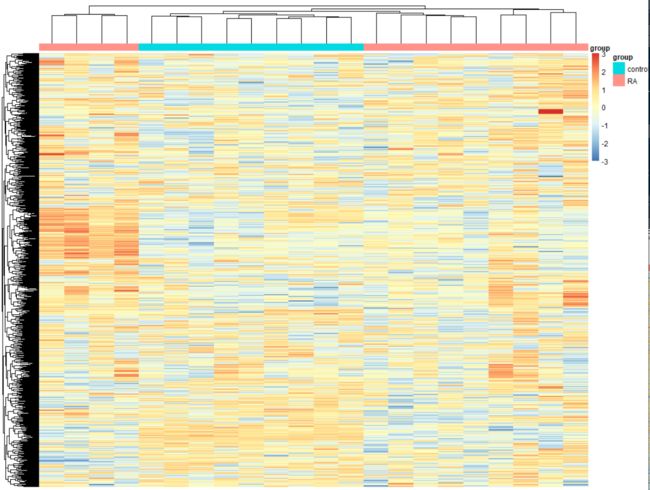
# 2.top 1000 sd 热图----
cg=names(tail(sort(apply(exp,1,sd)),1000))
n=exp[cg,]
# 直接画热图,对比不鲜明
library(pheatmap)
annotation_col=data.frame(group=Group)
rownames(annotation_col)=colnames(n)
pheatmap(n,
show_colnames =F,
show_rownames = F,
annotation_col=annotation_col
)
# 按行标准化
pheatmap(n,
show_colnames =F,
show_rownames = F,
annotation_col=annotation_col,
scale = "row",
breaks = seq(-3,3,length.out = 100)#规定取值对应颜色的而范围以及分1000个级别
#cluster_rows = TRUE 是否聚类
#cluster_cols = TRUE
)
dev.off()
四、差异分析
rm(list = ls())
load(file = "step2output.Rdata")
#差异分析,用limma包来做
#需要表达矩阵和Group,不需要改
library(limma)
design=model.matrix(~Group)
fit=lmFit(exp,design)
fit=eBayes(fit)
deg=topTable(fit,coef=2,number = Inf)
#为deg数据框添加几列
#1.加probe_id列,把行名变成一列
library(dplyr)
deg <- mutate(deg,probe_id=rownames(deg))
#2.加上探针注释,去除一个基因对应多种探针ID,随机去重
ids = ids[!duplicated(ids$symbol),]
#其他去重方式在zz.去重方式.R
deg <- inner_join(deg,ids,by="probe_id")
nrow(deg)
#3.加change列,标记上下调基因
logFC_t=1
P.Value_t = 0.05
k1 = (deg$P.Value < P.Value_t)&(deg$logFC < -logFC_t)
k2 = (deg$P.Value < P.Value_t)&(deg$logFC > logFC_t)
deg <- mutate(deg,change = ifelse(k1,"down",ifelse(k2,"up","stable")))
table(deg$change)
#4.加ENTREZID列,用于富集分析(symbol转entrezid,然后inner_join)
library(clusterProfiler)
library(org.Hs.eg.db)
s2e <- bitr(deg$symbol,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)#人类
#其他物种http://bioconductor.org/packages/release/BiocViews.html#___OrgDb
deg <- inner_join(deg,s2e,by=c("symbol"="SYMBOL"))
save(Group,deg,logFC_t,P.Value_t,gse_number,file = "step4output.Rdata")差异分析主要是用limma包来做的,通过lmFIT和ebays(用来拟合和校正t检验之中的方差部分),之后添加探针注释,标记上下调基因,加RNTREZID列来用于差异分析
五、差异分析后画图部分
rm(list = ls())
load(file = "step1output.Rdata")
load(file = "step4output.Rdata")
#1.火山图----
library(dplyr)
library(ggplot2)
#这次去重复是去除多个表达量对应一个基因的情况,随机去重
dat = deg[!duplicated(deg$symbol),]
## 规定横纵轴
p <- ggplot(data = dat,
aes(x = logFC,
y = -log10(P.Value))) +
geom_point(alpha=0.4, size=3.5,
aes(color=change)) +
ylab("-log10(Pvalue)")+
scale_color_manual(values=c("blue", "grey","red"))+
geom_vline(xintercept=c(-logFC_t,logFC_t),lty=4,col="black",lwd=0.8) +
geom_hline(yintercept = -log10(P.Value_t),lty=4,col="black",lwd=0.8) +
theme_bw()
p
#优秀的管道符号,将一个函数的运算结果传输到下一个函数中
for_label <- dat%>%
可以更换自己关注的基因名称
filter(symbol %in% c("HADHA","LRRFIP1"))##
# 火山图
volcano_plot <- p +
geom_point(size = 3, shape = 1, data = for_label) +
ggrepel::geom_label_repel(
aes(label = symbol),
data = for_label,
color="black"
)
volcano_plot
#2.差异基因热图----
load(file = 'step2output.Rdata')
# 表达矩阵行名替换
exp = exp[dat$probe_id,]
rownames(exp) = dat$symbol
if(F){
#全部差异基因
cg = dat$symbol[dat$change !="stable"]
length(cg)
}else{
#取前10上调和前10下调
library(dplyr)
dat2 = dat %>%
filter(change!="stable") %>%
arrange(logFC)
cg = c(head(dat2$symbol,10),
tail(dat2$symbol,10))
}
n=exp[cg,]
dim(n)
#差异基因热图
library(pheatmap)
annotation_col=data.frame(group=Group)
rownames(annotation_col)=colnames(n)
heatmap_plot <- pheatmap(n,show_colnames =F,
scale = "row",
#cluster_cols = F,
annotation_col=annotation_col,
breaks = seq(-3,3,length.out = 100)
)
heatmap_plot
#拼图
library(patchwork)
library(ggplotify)
volcano_plot +as.ggplot(heatmap_plot)
# 3.感兴趣基因的相关性----
library(corrplot)
g = deg$symbol[1:10] # 换成自己感兴趣的基因
g
M = cor(t(exp[g,]))
pheatmap(M)
library(paletteer)
my_color = rev(paletteer_d("RColorBrewer::RdYlBu"))
my_color = colorRampPalette(my_color)(10)
corrplot(M, type="upper",
method="pie",
order="hclust",
col=my_color,
tl.col="black",
tl.srt=45)
library(cowplot)
cor_plot <- recordPlot()
# 拼图
load("pca_plot.Rdata")
plot_grid(pca_plot,cor_plot,
volcano_plot,heatmap_plot$gtable)
dev.off()
#保存
pdf("deg.pdf")
plot_grid(pca_plot,cor_plot,
volcano_plot,heatmap_plot$gtable)
dev.off()