Chapter2 : Machine Learning Applied to the Modeling of Pharmacological and ADMET Endpoints
reading notes of《Artificial Intelligence in Drug Design》
文章目录
- 1.Introduction
- 2.Machine Learning Applied to ADMET Problems
-
- 2.1.The importance of a Favorable ADMET Profile
- 2.2.Data, Descriptors, Algorithms, Metrics
- 2.3.Data Are Key
-
- 2.3.1.Experimental Assay Data
- 2.3.2.Standardization of Chemical Structure
- 2.3.3.Preprocessing of Assay Data for Machine Learning
- 2.3.4.Examples for the Effort and Importance of Data Curation
- 2.4.Machine Learning Algorithms
-
- 2.4.1.History of Supervised ML Algorithms in Drug Discovery
- 2.4.2.Pros and Cons of Supervised ML Algorithms in Drug Discovery Industry
- 2.5.Descriptor
-
- 2.5.1.Molecular Descriptors
- 2.5.2.Atom Descriptors
- 2.6.Performance Metrics
- 2.7.Identification of Stable and Performant Models
- 2.8.Applicability Domain
- 2.9.Models for complex and Multiple Endpoints
-
- 2.9.1.Modeling Physicochemical ADMET Endpoints with Multitask Graph Convolutional Networks
- 2.9.2.Modeling of in Vivo Endpoints
- 2.9.3.Modeling of Drug Metabolism
- 2.10.Application Examples
-
- 2.10.1.Bayer's integrated ADMET Platform
- 2.10.2.Guiding the Design of Combination Libraries
- 2.10.3.Combing Cheminformatics and Physics-Based Methods in Lead Optimization
- 3.Summary and Outlook
1.Introduction
- The pharmacological activity on the primary target, together with absorption, distribution, metabolism, excretion and toxicity (ADMET) are the main parameters for the discovery and optimization of new drugs.
- Both the structure-based and the machine learning-based approaches can apply descriptions of the molecules and their interactions derived empirically or physics-based.
- By far the most commonly applied QM methodology in industry is density functional theory (DFT), mainly due to its beneficial cost-accuracy ratio.
- Substantial advances in automation, methodology, and computing power have enabled the increasing adoption of molecular dynamics and accurate free energy calculations in industry.
2.Machine Learning Applied to ADMET Problems
2.1.The importance of a Favorable ADMET Profile
- Kola and Landis in 2004 showed that in the period between 1991 and 2000 the attrition rates due to PK and bioavailability went down significantly (from 42 to 10%), whereas in the same period attrition due to toxicology and clinical safety significantly increased, attributing to an increase in size and lipophilicity of the compounds.
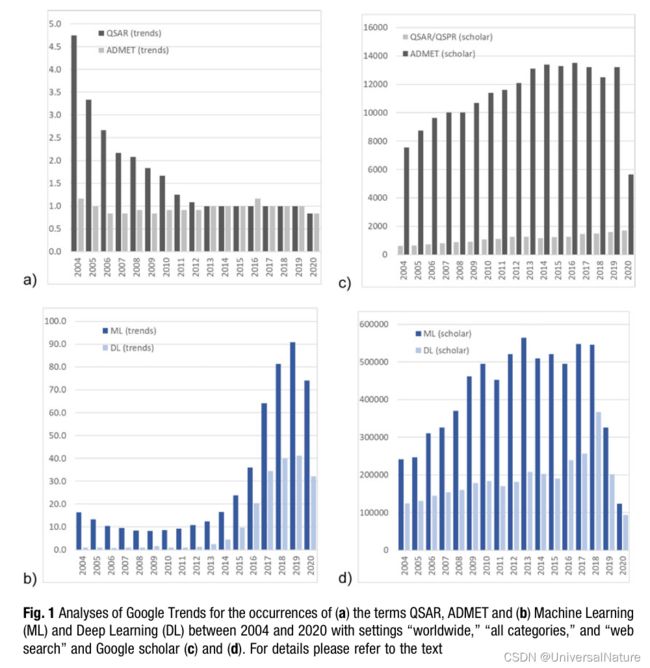
2.2.Data, Descriptors, Algorithms, Metrics
- We discuss three key ingredients to stable and predictive models, namely data, descriptors and algorithms, as well as the metrics applied to identify those.
2.3.Data Are Key
- There are two types of data, namely the chemical structures and assay data that are learned.
2.3.1.Experimental Assay Data
-
Historically, assay definitions and upload procedures were often setup in a way to allow direct consumption of the data by the requesting research project, but not with further usage in mind.
-
In March 2016 a publication appeared by a consortium of scientists that outlined four foundational principles—Findability, Accessibility, Interoperability, and Reusability—abbreviated as FAIR principles, that describe procedures to a FAIRification process.
-
Close communication with experimentalist is of utmost importance for the data scientist in data preprocessing state.
-
An assay is composed of four components: a biological or physicochemical test system, a detection method, the technical infrastructure, and finally data analysis and processing.
- biological systems have a certain variability, like differences in cell activity
- detection method has a certain variability and in case of UV is highly dependent on the molecules
- stickiness of highly lipophilic compounds to glass or plastic parts like pipettes reduces the available concentration of the compound compared to the apparent concentration, resulting in erroneously too high values
2.3.2.Standardization of Chemical Structure
- A study by Young et al. on six public and private databases gave error rates ranging from 0.1 to 3.4% and proved that including erroneous results in significant drops model accuracies.
- The utmost goal here is to come up with a standardization protocol, which is applied not only during model training but also for model application.

-
It might be necessary to cleave off the leaving groups of prodrugs in case the experimental property is determined for the pharmacologically active substance.
-
Inconsistent hydrogen treatment may result in differing descriptor values.
-
Most of the descriptor packages cannot cope with stereochemistry anyhow, and therefore stereocenters are flattened.
-
One may, in case of modeling of target affinity profiles, additionally apply structure filters on frequent hitters like PAINS or “Hit Dexter” to avoid noise due to unspecific binding data.
-
the European Union-funded consortium IMI MELLODDY as part of the innovative medicine initiative (IMI) has developed and published an end-to-end open source tool for the process described under the name MELLODDY_tuner . The tool is used to standardize the data needed for the project to succeed in the endeavor of federated and privacy-preserving machine learning to leverage the world’s largest collection of small molecules with known biochemical or cellular activity to enable more accurate predictive models and increase efficiencies in drug discovery.
2.3.3.Preprocessing of Assay Data for Machine Learning
-
Combination of data from different sources poses further challenges, an alternative is to establish a multitask ML model that predicts the values of one assay and uses the other variant as a helper task.
-
There are three categories of data that require curation:
- data with attached comments, some comments such as “not fully dissolved or calibration issue” allowed to filter out experiments that not trustworthy.
- censored data is data which is outside the detection window or the serial dilution window. For classifier models, such data can be used, but for numerical models they have to removed. Intermediate censored data should always be removed.
- structures with multiple test values including outliers, there are different approach to solve this problem such as removal or replace it with the median value (rather than mean value).
2.3.4.Examples for the Effort and Importance of Data Curation
- There are two examples on the high effort needed for data curation: Bayer’s pKa model and author’s SoM model.
2.4.Machine Learning Algorithms
2.4.1.History of Supervised ML Algorithms in Drug Discovery
- The basic principles and limitation of ML algorithms used in today’s drug discovery have been described by Mitchell and by Lo et al. .Furthermore, a very systematic study of ML algorithms applied in chemical health and safety has recently been published.

2.4.2.Pros and Cons of Supervised ML Algorithms in Drug Discovery Industry
-
Random Forests have long been the method of choice in Bayer’s ADMET platform for several reasons:
- in combination with circular fingerprints, the RF model performance was generally highest and very robust
- out-of-the-box hyper-parameters that determine the configuration of the RF algorithm are usually optimal and do not need to be search and optimized as is i.e. the case for SVMs
- ensemble models such as random forests bring along can be used as a measure of confidence of the individual prediction
-
The advantages of deep neural networks in Drug Discovery appear to be that
- there is no need for feature engineering e.g. graph-convolutional networks
- neural network input can be utilized rather flexible such as different format data or confidential data
2.5.Descriptor
- Any representation of a molecule that is used for computational chemistry will always be some abstraction with some loss of information.
2.5.1.Molecular Descriptors
-
Here are classification scheme from Wikipedia, which defines five main classes:
- 0D-descriptors (i.e. constitutional descriptors, count descriptors). They are highly correlated which further reduces the information content as shown in Table 1 that provides the Pearson correlation coefficients for the Lipinski rule-of- five and the Veber properties for randomly picked 1% of the Bayer compound deck.

-
1D-descriptors (i.e. list of structural fragments, fingerprints). Our work-horse descriptors for more than a decade, confirmed by many publications, are circular extended connectivity fingerprints (ECFP), which encode properties of atoms and their neighbors into a bit vector of certain topological (numbers of bonds to starting atom) radius and feature type (element, function as donor, acceptor, etc., atom type).
-
2D-descriptors (i.e. graph invariants). Graph invariant 2D-descriptors like topology or connectivity indices at least in our hands often yield overfitted models that work well in cross-validation but are not predictive on external test sets.
-
3D-descriptors (such as, for 3D-MoRSE descriptors, WHIM descriptors, GETAWAY descriptors, quantum-chemical descriptors, size, steric, surface and volume descriptors). Main issue is their dependence on the conformation which introduces ambiguities and noise.
-
4D-descriptors (such as those derived from GRID or CoMFA methods, Volsurf). These approaches have the additional limitation of being dependent on the alignment of the ligands which is sometimes not obvious and only possible in the case of congeneric series.
-
There are now public databases and model repositories on compound collections such as QsarDB, Danish (Q)SAR Models database, QSAR toolbox etc.
-
Actually, the perception of learning the optimal representation directly from the molecule is not exactly correct, because the SMILES or InCHi typically used as structure input is already an abstract representation (i.e. a reduction) of the molecule.
-
Winter et al. applied the autoencoder-autodecoder concept to learn a fixed set of continuous data-driven descriptors, the CDDD, by transforming random SMILES to canonical SMILES during training. The resulting descriptor is based on approximately 72 million compounds from the ZINC and PubChem databases. The validity of the approach was tested by model performance on eight QSAR datasets and by application to virtual screening. It showed similar performance to various human-engineered descriptors and graph-convolutional models.
2.5.2.Atom Descriptors
-
Machine learning problems that are concerned with the reactivity of atoms like reaction rates and regioselectivity, pKa values, the prediction of the metabolic fate, or hydrogen bonding interactions require encoding of the properties of the atoms and their surrounding into specialized atom descriptors. In many applications the descriptor values are directly retrieved from quantum chemical calculations.
-
There are also examples of well-performing classical neighborhood encoding atom descriptors for SoM prediction and regioselectivity in Diels-Alder reactions.
2.6.Performance Metrics
-
Common metrices for regression models include R2, root mean square error (RMSE), and Spearman’s rho
- R2 is the coefficient of determination and gives information on how close the data fits the regression line. It’s might be necessary to calculate the R2 for just the relevant value range of the predicted property and not for the full range.
- The RMSE is the standard deviation of residuals and indicates how close the predicted values are to the real data points and is a reliable, general error metric.
- Spearman’s rho is a nonparametric rank correlation coefficient. For a perfect ordering of the predictions from low to high in accordance with experimental values, rho will be 1. It will reduce for each misordered pair of objects and for perfect inverse ranking will be -1. A high value for rho indicates that the model applied in a project will be able to answer the question.
-
Common metrices for assessing the quality of classification models are derived from the Confusion Matrix, also named Contingency Matrix.
-
In case of highly imbalanced datasets, the accuracy can be misleading e.g. if the model always predicts the higher populated class, it will get a high accuracy without being predictive.
-
Specificity or true negative rate, is the proportion of observed negatives that are predicted as such while sensitivity, also called true positive rate or recall, is the proportion of observed positives that are predicted correctly. Another metric focusing more on the predictions than the observed values is the positive predictive value, also called precision, which shows the proportion of correctly predicted positives out of all predicted positives. F-Score which is the harmonic mean of precision and sensitivity.
-
The Matthews correlation coefficient (MCC) is the geometric mean of the regression coefficient and is also suitable for classification problems with imbalanced class distributions.
-
Cohen’s kappa is also a good measure that can handle imbalanced class distributions and shows how much better the classifier is compared to a classifier that would guess randomly according to the frequency of each class.
-
Another popular metric is the receiver operation characteristic (ROC) graph to visualize the performance of the classification algorithm. The area under the ROC curve (ROC AUC) is the numerical metric used to describe the ROC curve.
2.7.Identification of Stable and Performant Models
-
The process accepted as best practices for machine learning which developed over the last 20 years and that is now generally applied is described in current reviews and outlined in detail in the respective OECD guideline.
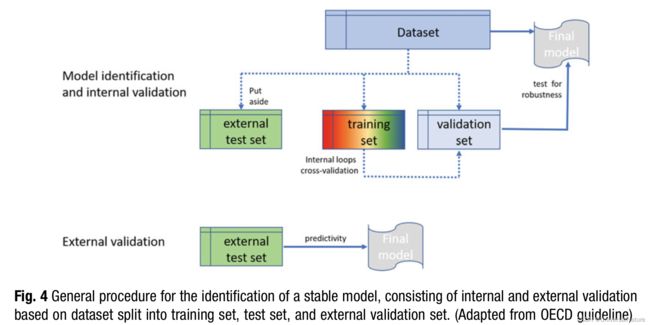
-
Validation strategies broadly applied are cross-validation, bootstrapping and Y-scrambling.
2.8.Applicability Domain
- The region of Chemical space where the model’s predictions are reliable is limited in comparison to the immense drug-like space and the prediction accuracy for completely novel molecules may in practice be disappointing.

- Many different so-called applicability domain (AD) measures have been introduced in recent years, which can be grouped into two classes:
- methods that apply distance measures on how well the future object is embedded in the training set, are termed “novelty detection”, it can be applied for any algorithms.
- methods that quantify the distance to the decision boundary of the classifier are called “confidence estimation”,it can’t be applied for any algorithms, but it is superior in general.
2.9.Models for complex and Multiple Endpoints
2.9.1.Modeling Physicochemical ADMET Endpoints with Multitask Graph Convolutional Networks
-
By sharing of parameters in (some of) their hidden layers between all tasks multitask neural networks force the learning of a joint representation of the input that will be useful to all tasks.
-
The main advantages of multitask learning are
- regularization, in that the model has to use the same amount of parameters to learn more tasks
- transfer learning, whereby learning-related tasks help extracting features that are useful in a more general way
- dataset augmentation, by combining smaller tasks with larger tasks to avoid overfitting on the small task
-
Multitask effect are highly dataset-dependent which suggests the use of dataset-specific models to maximize overall performance.
-
The work of author is that the multitask graph convolutional network performed on par or better than the single-task graph convolutional network and outperformed single-task random forests or neural networks with circular fingerprint descriptors, especially in the case of solubility, where the improvement was break-through.
2.9.2.Modeling of in Vivo Endpoints
- Oral bioavailability F is defined as the extent of the oral dose that is available to produce pharmacological actions. It is defined as the ratio of the dose-normalized exposures after oral (po) versus intravenous (iv) administration. Exposure is determined as the area under the curve from multiple blood plasma samples taken over a period of typically 24h.
- For single compounds, physiological based pharmacokinetic modeling (PBPK), a methodology that describes the body of the species as interacting compartment allows to describe the time-dependent exposure in different organs depending on additional parameters like body weight, gender, disease state at a quality that can for instance be used to in silico determine pediatric doses or risks.
2.9.3.Modeling of Drug Metabolism
-
Any oral drug first passes the liver before entering the rest of the body. Metabolic transformations occur in two phases. In phase I, mostly cytochrome P450 enzymes increase polarity by oxidative and reductive reactions. In phase II, a plethora of enzymes like UDP-glucuro-nosyltransferase, sulfo-transferases, or glutathione S-transferases conjugate specific fragments to the phase I metabolites for renal excretion.
-
The high effort and limitation in experimental assay capacity for the identification of SoMs has led to many computational approaches over the last 20 years, applying docking, molecular dynamics, quantum chemistry calculations, and machine learning, with and without incorporation of protein target information. The reader is referred to Kirchmair et al. for a comprehensive overview over experimental and computational approaches.
-
The lability of atoms with regards to metabolic reactions is determined by their chemical reactivity, i.e. the local electron density and the steric accessibility of the respective atoms, necessitating atomic descriptors instead of molecular ones, as well as machine learning for atoms instead of molecules.
2.10.Application Examples
2.10.1.Bayer’s integrated ADMET Platform
- The two last but most important steps to take in end are
- to make the models accessible to the users in an easy-to-use platform
- to constantly communicate to and train the users
- The portfolio of models and their quality increased constantly over the years, as indicated in Fig. 6. With this, the effort for manual model retraining became more and more prohibitive, and in parallel we and others found that regular m
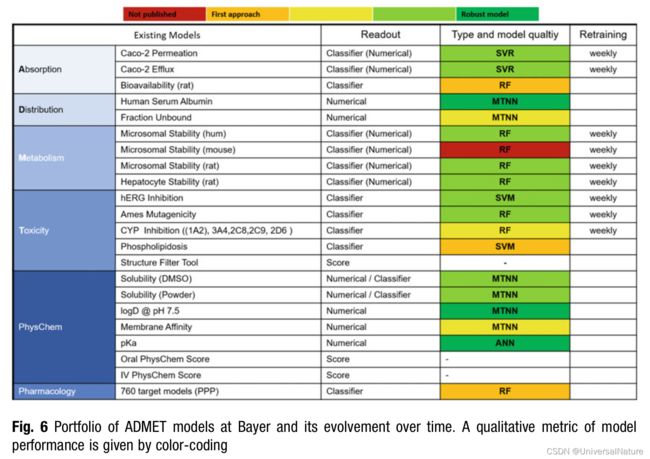
odel retraining has a positive effect on the performance in active projects, even when only adding 20–50 compounds per interval.

2.10.2.Guiding the Design of Combination Libraries
- In parallel to computational approaches to generate new chemical matter as starting points for drug discovery projects like virtual screening or de novo design, high-throughput screening is still a valuable tool to identify hits. Experimental testing nevertheless erodes the chemical library twofold, by substance consumption and by novelty erosion, since any hit will then indirectly expose a certain subset of the compound space.
2.10.3.Combing Cheminformatics and Physics-Based Methods in Lead Optimization
-
The complex multi-parameter optimization necessary to find the best compromise of many optimization parameters is a key challenge in drug discovery projects.
-
In this section, we sketch a project situation from 2016, where we exemplarily show how to tackle the prioritization of compounds from a large virtual chemical space by combining cheminformatics and physics-based approaches.
3.Summary and Outlook
-
The willingness to share more of those data in conjunction with the use of block-chain technologies enables the privacy preserving exchange of data among many pharmaceutical companies, increasing the data basis for models by several orders of magnitude.
-
Some of those machine learning models have reached the quality to significantly reduce or halt experimental measurements. However, this is not valid for all endpoints and despite the availability of large homogenous datasets some ADMET endpoints can still not be modeled with sufficient quality. Phys-chem/ADMET properties as well as chemical synthesizability are mainly modeled with data-based approaches such as machine learning.
-
For pharmacological endpoints, the data are sparse and only a smaller fraction (typically <30%) of a larger diverse drug target portfolio will be covered by ML models with sufficient predictivity. Pharmacological activity is often addressed with protein-structure based approaches.