PySCENIC(三):pyscenic单细胞转录因子分析可视化
更多精彩内容请至我的公众号---KS科研分享与服务
先加载需要的R包,都加载了,没毛病。
setwd("/home/shpc_100828/Pyscenic/")
#加载分析包
library(SCopeLoomR)
library(AUCell)
library(SCENIC)
#可视化相关包,多加载点没毛病
library(dplyr)
library(KernSmooth)
library(RColorBrewer)
library(plotly)
library(BiocParallel)
library(grid)
library(ComplexHeatmap)
library(data.table)
library(ggplot2)
library(pheatmap)
将loom文件读入R,提取数据。
sce_SCENIC <- open_loom("sce_SCENIC.sce_SCENIC")
# exprMat <- get_dgem(sce_SCENIC)#从sce_SCENIC文件提取表达矩阵
# exprMat_log <- log2(exprMat+1) # log处理
regulons_incidMat <- get_regulons(sce_SCENIC, column.attr.name="Regulons")
regulons <- regulonsToGeneLists(regulons_incidMat)
class(regulons)
regulonAUC <- get_regulons_AUC(sce_SCENIC, column.attr.name='RegulonsAUC')
regulonAucThresholds <- get_regulon_thresholds(sce_SCENIC)
第一个可视化:
RSS分析,查看细胞类型特异性转录因子,需要先加载seurat对象,提取metadata信息,并进行分析!默认是点图!
human_data <- readRDS("~/Pyscenic/human_data.rds")
cellinfo <- [email protected][,c('celltype','group',"nFeature_RNA","nCount_RNA")]#细胞meta信息
colnames(cellinfo)=c('celltype', 'group','nGene' ,'nUMI')
######计算细胞特异性TF
cellTypes <- as.data.frame(subset(cellinfo,select = 'celltype'))
selectedResolution <- "celltype"
sub_regulonAUC <- regulonAUC
rss <- calcRSS(AUC=getAUC(sub_regulonAUC),
cellAnnotation=cellTypes[colnames(sub_regulonAUC),
selectedResolution])
rss=na.omit(rss)
rssPlot <-
plotRSS(
rss,
zThreshold = 3,
cluster_columns = FALSE,
order_rows = TRUE,
thr=0.1,
varName = "cellType",
col.low = '#330066',
col.mid = '#66CC66',
col.high = '#FFCC33')
rssPlot
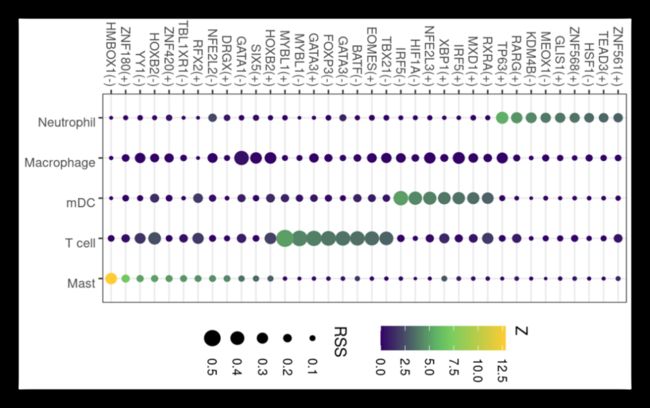
image.png
我们也可以提取数据,用热图的方式呈现,这里我是用ggheatmap做的,也可以用pheatmap、complexheatmap或ggplot2做。
rss_data <- rssPlot$plot$data
devtools::install_github("XiaoLuo-boy/ggheatmap")
library(ggheatmap)
library(reshape2)
rss_data<-dcast(rss_data,
Topic~rss_data$cellType,
value.var = 'Z')
rownames(rss_data) <- rss_data[,1]
rss_data <- rss_data[,-1]
colnames(rss_data)
col_ann <- data.frame(group= c(rep("Neutrophil",1),
rep("Macrophage",1),
rep("mDC",1),
rep("T cell",1),
rep("Mast",1)))#列注释
rownames(col_ann) <- colnames(rss_data)
groupcol <- c("#D9534F", "#96CEB4", "#CBE86B", "#EDE574", "#0099CC")
names(groupcol) <- c("Neutrophil","Macrophage","mDC", "T cell","Mast")
col <- list(group=groupcol)
text_columns <- sample(colnames(rss_data),0)#不显示列名
p <- ggheatmap(rss_data,color=colorRampPalette(c('#1A5592','white',"#B83D3D"))(100),
cluster_rows = T,cluster_cols = F,scale = "row",
annotation_cols = col_ann,
annotation_color = col,
legendName="Relative value",
text_show_cols = text_columns)
p
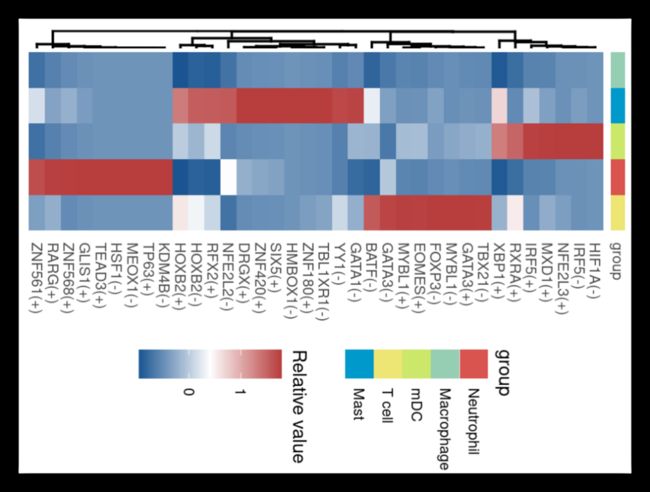
image.png
第二个可视化:
将转录因子分析结果与seurat对象结合,可视化类似于seurat!
next_regulonAUC <- regulonAUC[,match(colnames(human_data),colnames(regulonAUC))]
dim(next_regulonAUC)
regulon_AUC <- regulonAUC@NAMES
[email protected] = cbind([email protected] ,t(assay(next_regulonAUC[regulon_AUC,])))
#自己选定感兴趣的或者比较重要的转录因子,这里我是随机的
TF_plot <- c("ZNF561(+)","FOXP3(+)","YY1(+)","HOXB2(+)",
"TBX21(+)","TCF12(+)","STAT2(+)","SOX21(+)",
"RBBP5(+)","NR2F6(+)","NELFE(+)","MAFG(+)")
DotPlot(human_data, features = TF_plot)+
theme_bw()+
theme(panel.grid = element_blank(),
axis.text.x=element_text(hjust =1,vjust=1, angle = 45))+
labs(x=NULL,y=NULL)+guides(size=guide_legend(order=3))

image.png
上面我们展示的是转录因子在不同细胞的评分,按照这个道理,我们依然可以选定某种细胞,看样本间转录因子的差别!
DotPlot(human_data, features = TF_plot, group.by = 'group')+
theme_bw()+
theme(panel.grid = element_blank(),
axis.text.x=element_text(hjust =1,vjust=1, angle = 45))+
theme(legend.direction = "horizontal",
legend.position = "bottom")+
labs(x=NULL,y=NULL)
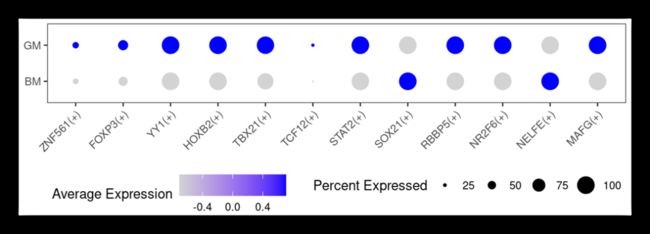
第三个可视化:
展示转录因子平均活性!
cellsPerGroup <- split(rownames(cellTypes),
cellTypes[,selectedResolution])
regulonActivity_byGroup <- sapply(cellsPerGroup,
function(cells)
rowMeans(getAUC(sub_regulonAUC)[,cells]))
regulonActivity_byGroup_Scaled <- t(scale(t(regulonActivity_byGroup),
center = T, scale=T))
regulonActivity_byGroup_Scaled=na.omit(regulonActivity_byGroup_Scaled)
hm <- draw(ComplexHeatmap::Heatmap(regulonActivity_byGroup_Scaled, name="Regulon activity",
row_names_gp=grid::gpar(fontsize=6),
show_row_names = F))
hm #可视化所有的TF
当然了,全部展示没有啥意义,还是可以提取数据,可视化需要的TF!