转录组RNA-seq分析前沿进展综述
RNA测序(RNA-seq)已经成为分析基因差异表达和mRNAs差异剪接不可或缺的工具。随着下一代测序技术的发展,RNA-seq也在发展。
目前,RNA-seq方法可用于研究RNA生物学的许多不同方面,包括单细胞基因表达、翻译和RNA结构。随着直接RNA-seq技术和更好的数据分析工具的出现,RNA-seq的发展有助于更全面地理解生物科学,本文解读一篇2019年发表在Nature的RNA-sequencing综述。

DGE:基因差异表达
流程 :
RNA提取→mRNA富集或rRNA耗尽→cDNA合成→适配器连接→测序文制备→ 高通量平台(通常是Illumina)上进行测序,每个样本的读深度为1000万-3000万。
将测序读数对齐组合到一个反义读数→量化与转录本重叠的读数→样本之间的过滤和归一化→构建样本组之间单个基因或转录本表达水平的显著变化的统计模型。 早期的RNA-SEQ实验从大量组织中产生了DGE数据,并证明了它在广泛的生物和系统中的应用。
RNA-SEQ经常被用作不同的生物学应用,DGE分析(差异表达分析)仍然是RNA-SEQ的主要应用。
本文中,首先介绍了DGE的“基本准则”短读RNA-SEQ分析,然后将标准短读方法与新兴的长读RNA-SEQ和dRNA-SEQ技术进行比较。描述了短读排序建库的发展、实验设计和工作流。还有单细胞测序和空间分辨转录组分析。以及转录和翻译动力学、RNA结构、RNA-RNA和RNA-蛋白质相互作用的分析。 最后,讨论了RNA-SEQ未来可能的发展前景,单细胞测序和空间RNA-SEQ方法是否会像DGE分析一样成为常规?RNA-SEQ分析的长读可能在什么领域取代短读?

短读测序技术
目前短读测序技术已经成为常用的转录组检测方法,主要的核心步骤:RNA提取→cDNA合成→接头连接→PCR扩增→测序分析。由于片段化产生的cDNA长度通常小于200bp。例如illumina技术
长度测序技术
能够对单个RNA分子进行测序,减少序列测定中的歧义性,降低假阳性率。例如pacbio技术
长读直接测序技术
dRNA-seq,直接用RNA进行测序,消除逆转录过程的偏差,产生超过100bp的读取长度。
优点:改进了异构体检测性能、可用于估计polyA尾长。

比较长读和短读技术
- 长读:缺点是通量低、错误率高。优点是能够读取单个转录本。
- 短读:优点是通量高,测序规模大,差异基因表达敏感性强。
长读测序的一个重要的问题——错误率比较高,如果每个样品被单独多次测序,再通过计算处理成一致序列,那么分子测序的次数的越多,错误程度越低。 长度测序的灵敏度受到三个因素影响:长RNA分子以全长转录组形式存在,即使低水平的RNA降解也会限制测序;文库制备技术限制,常用的逆转录酶无法满足实验需求;测序平台发展限制。
改进构建文库方法
RNA-seq最初用于分析聚腺苷酸转录本,方法源自于表达序列标签和微阵列研究。然而这些技术具有一定局限性。
- cDNA合成前对RNA进行片段化处理,提升丰度估计的准确性。
- 链特异性文库,测序时利用oligo-dT富集、UMIs特异性标注,利于实验分析。
富集poly(A)尾巴
多数转录组测序结果都是从oligo-dT富集mRNA中获得,它含有多聚poly A的尾巴,测序的重点在转录组的蛋白质编码区。
许多非编码RNA(比如miRNA和enhancer RNA)不是多聚核苷酸,因此无法用该法研究。我们可以选择用oligo-dT法或者rRNA降解法(WTA),前者不能获取短的非编码RNA,后者需要特定的miRNA.
WTA通过编码和一些非编码RNAs产生RNA-seq数据,它与降解样品相容,导致poly A和转录本分离,rRNA的去除通过RNase H酶特异性实现。 oligo-dT和rRNA降解法(WTA)都可以用于DGE研究,后者可以检测到更多转录本,但是贵!
富集3'端用于标记和选择
短读测序需要每个样本1000万-3000万的读数才能进行高质量的DGE分析,对于资源有限的朋友来说,可以考虑用3'-tag计数,降低成本同时得到更多数据。
每个转录本从3'端开始生成一个片段,标签的丰度与RNA浓度呈正比,这称为标签测序协议。
富集转录起始位点5'末端
TSS:转录起始位点
该法利用生物素化模板转换寡聚核苷酸产生cDNA,然后在5'端附近片段化,产生短cDNA标签。如果只用该法会产生大量假阳性TSS峰,建议使用正交法进行验证确认。
应用分子标识符检测PCR重复
RNA-seq数据通常具有很高的重复率,许多读数映射到转录组的同一位置。
在全基因组测序中,重复读数代表PCR失误而被删除。而转录组测序中,重复代表真实的生物信号被保留。后者需注意只有一对片段的一段必须相同,才能确定其为重复序列。
UMIs:独特分子识别符,在文库制备过程中添加的短序列,其直接与RNA连接。
UMIs改善数据分析的错误率,放大偏差,消除可能导致等位基因频率计算错误的人工扩增制品,其正成为单细胞测序文库制备的标准工具之一。
改进降解RNA分析方法
文库制备的发展改善了对低质量或降解RNA的分析,以往的低质量RNA会导致基因覆盖不均匀、DGE假阳性高、重复率高。转录组文库构建可以用来降低RNA降解的影响。
设计更好的转录组实验
实验的设计对结论和数据至关重要,需要考虑复制次数、测序读取深度、单末端或者双末端读取的选择。
确定复制次数
足够多的生物复制能够获得更加精确的信息,解读生物变异性,任何高通量RNA实验都必须以复制的方式进行,在确定最佳复制次数时应该考虑到:效应大小、组内变异、预期假阳性率、最大样本量等。
确定正确的复制次数并不容易,对于高度多样化的样本,需要更多的复制,以确定变化规律。
确定读取深度
建库完成后,需要决定进行多深的测序?读取深度指每个样本获得的序列读取目标数目。真核生物DGE实验每个样本大约需要1000万-3000万左右。每个样本的读数能提供转录本丰度估计值,如果有足够的复制次数,那么较少的测序即可解决实验问题。
选择合适的参数
- 读取长度越长,测序DNA覆盖程度越高。对于定量分析如DGE时,长读没有太大作用。但对定性分析如异构体,则可能有帮助。
- 单末端读取和双末端读取也相类似,前者每个cDNA片段只产生一个序列,后者产生俩。若需要更多核苷酸覆盖度分析,长读和双末端测序是首选!DGE分析只需要计算读数映射,单末端测序即可确定大部分基因的源头,另外,双末端测序可以帮助解决映射歧义问题。
RNA-seq数据分析流程
↓ ↓ ↓转录组差异表达分析常用软件工具
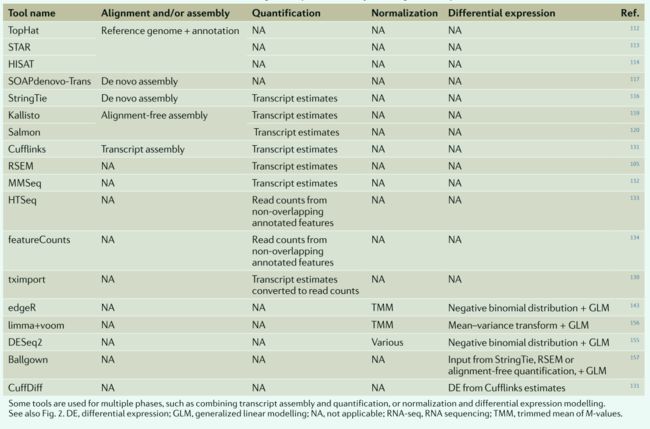
分析序列读数的方法非常多,不同的工具和方法对结果产生不同影响,使用最佳的工具能帮助我们解决更多问题。DGE差异表达分析常用四个步骤,分别是: 对齐、组装、量化、归一化、构建模型。
第一步:序列对齐与组装
测序产生fastq格式原始文件,首先将序列映射到已知的转录组或者基因组,这个过程使用TopHat,STAR,HISAT等工具完成。测序的cDNA来自RNA,因此可能跨过外显子边界,执行剪切对比时,允许读数间隙。
如果没有高质量基因组注释可用或者希望映射到转录本时,可以使用StringTie,SOAPdenovo-Trans工具。
-
流程A:hisat→htseq→TMM→edgeR
-
流程B:Kallisto→TXI→TMM→DESeq2
-
流程C:tophat→cufflinks→cuffdiff2

tips:如果基因组注释缺失或者不完整时,选择从头组装的工具。
新软件:Sailfish118, Kallisto119,Salmon【计算效率高、免费、直接映射不需要单独量化步骤、适用于较长的转录本、在低丰度转录本方面不准确】
第二步:转录本丰度量化
一旦序列读数被映射到转录组或基因组的位置上,即可将其分配到基因组或转录本确定丰度。量化采用的方法会对结果产生重大影响,利用转录组注释信息与已知的基因重叠信息进行计数,得出单个基因所有转录本异构体的丰度信息。
短读序列不会跨过剪接链接,因此不能分配明确的异构体,不同基因长度的异构体之间差异表达可能导致更准确的结果。
常用的工具:e RSEM,CuffLinks,MMSeq,HTSeq同源和重叠的转录本可能从分析中排除。
表达矩阵:Express Matrix
每一行是表达特征(基因或者转录本) 每一列是一个样本 值是实际读数或者估计丰度
第三步:过滤和归一化
通常,量化之后的读数也需要进行过滤和归一化,用来说明读取深度、表达模式、技术偏差之间的差异。
- 过滤:去除低丰度重复、改善检测结果
- 归一化:规范表达矩阵,转化丰度量,校正细微差异
尖峰控制:在处理前添加已知浓度的外源核酸序列,它们能够以不同浓度分布,用于检测反应效率和误差假阳性校正。
归一化的两个关键假设:
- 多数基因表达水平在复制组间保持稳定
- 不同样本在总mRNA水平没有显著差异
若以上两个假设不成立,则需要借助TMM均值修剪法,包括edgeR差异表达分析来弥补。 选择合适的归一化方法:尝试运用多种方式进行分析,对比结果的一致性,若差异较大,应进一步探索,找出差异来源。
另外一种解决方式是尖峰控制RNAs:在文库制备过程中,预先引入合适的外来RNA序列,尖峰插入RNA变异体,由于尖峰的浓度已知,读数和浓度呈正相关,因此可以用于校准样本的表达水平。
但是,实际过程中,在预定水平一致的插入尖峰很困难,在基因层面的读数比转录层面更可靠,因为异构体可以在样本中以不同的浓度表达。目前,尖峰控制并没有广泛使用,少量应用于单细胞测序领域。
第四步:差异表达建模
当序列被处理成表达矩阵后,实验即可被建模,从而确定哪些复制信息可能影响到表达水平。一些模型读取基因水平表达的计数,另一些模型依赖于转录水平的估计。前者以利于对齐的计数,并使用广义线性模型来进行评估。
工具:edgeR, DESeq2 and limma+voom 建模差异亚型表达量的工具:CuffDiff,MMSEQ,Ballgown。需要更多的计算量,结果也有较大变化。但是实际上前期的对齐、过滤、归一化等步骤对结果的影响更大!
超体RNA分析
标准的RNA seq分析是利用大量组织或者细胞进行的,不能保存空间三维信息和细胞类型,不利于我们理解生物的复杂性。为了能深入研究,大佬们提出了超RNA分析技术,如单细胞测序和空间转录组等。
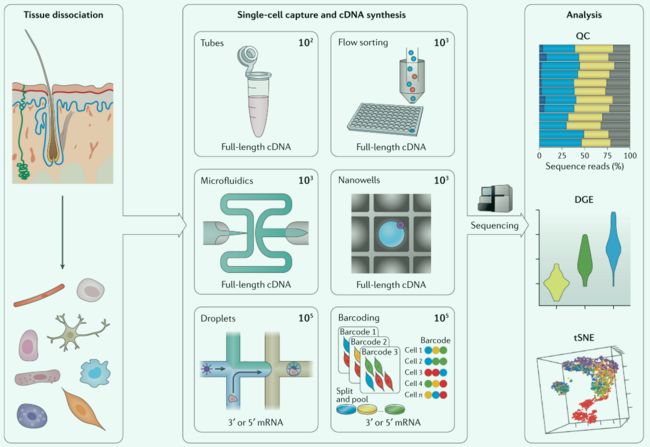
单细胞测序
scRNA-seq以单个细胞为样本(使用流式细胞仪,需要分离细胞,操作难度大),然后标记扩增其RNA,过程与常规转录组测序相似。单细胞分离后,裂解释放rna进行cDNA的合成,并制备文库。在制备文库过程中,单个细胞的rna用pcr扩增,引入了pcr偏差,可以用UMIs(分子标识符)进行校正。虽然只有少数转录本会被逆转录,但泊松分布法取样限制了检测的灵敏度,能够产生可用数据。
一般单细胞测序时,需要考虑以下问题:
- 是否沿着转录本全长读取?
- 分析更多细胞(广度)还是分析每个细胞更多转录本(深度)?
- 实验成本如何?
单细胞测序的特点:
吞吐量较低,每个细胞都需要独立处理,允许用户选择性剪接和等位基因特异性表达,推断异构体的能力较差,需要从广度和深度两个方向考虑问题。
常用方法:平板或微流控制法(捕获少量细胞,但检测的基因多,形成单个数据集)
未来,单细胞测序可能会像今天转录组测序一样批量使用。。。
空间转录组测序
目前,转录组测序提供了详细的数据,但是无法获取空间信息,这是研究过程中的一大难题。空间组学的两种方法:
空间编码和原位转录组学
空间编码:通过激光捕获显微切割的方法,从组织切片中获取RNA的空间位置编码信息。
原位转录组学:对细胞中RNA进行测序或者成像从而产生组织切片的数据。
目前需要解决的两个问题:
- 改法只能用于新鲜组织
- 分辨率有限制
上述技术不成熟,也有一些替代方法:单分子荧光原位杂交和基于成像法,能够产生更窄的图谱,直接检测RNA,同时提供组织结构和微环境信息。
超稳态RNA分析
DGE分析RNA-seq测量稳定的mRNA水平(通过平衡转录、加工、降解来维持),也可以用于转录和翻译过程和动力学研究。
新生RNA:Nascent RNA,指刚刚转录完成,未经加工运输的RNA。
用新生RNA测量活性转录
基因表达是一个动态过程,DGE在测量复杂快速变化时受到限制,RNA-seq能够用于TSSs图谱和新生RNA定量,从而使rna动力学研究成为可能。 新生RNA法特点:半衰期短、丰度低
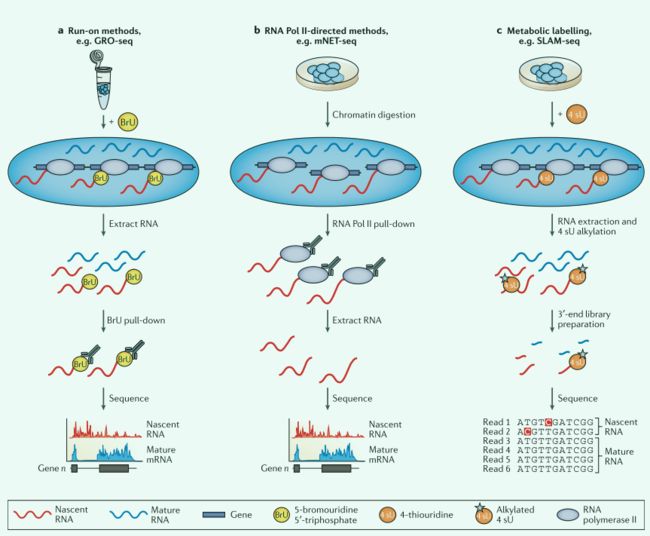
原生伸长转录测序:native elongating transcription sequencing (NET- seq),缺乏特异性,可能会污染新生RNA富集。
4-Thiouridine:4-硫代吡啶,真核生物核酸中天然不存在,容易结合,因此可用于初生RNA分析。进行代谢脉冲标记能够识别新生RNA,缺点是:时间长、灵敏度低。
初生RNA的分析方法受到非特异性背景和降解RNA的负面影响,标记比较复杂,限制了灵敏度,进一步的发展可能与空间组学结合,或者RNA-蛋白质互作结合的方法。
核糖体图谱法测定活性翻译
RNA-seq重点关注样品中的mRNA种类和数量,但mRNA不一定直接对应蛋白质的产生。以下两种方法帮助理解“翻译体(从mRNA转化而来的完整蛋白质集合。
- 多核糖体分析法
- 基于RNA-seq的核糖体足迹法
上述两种方式能得知有多少个核糖体占据了一个转录本?以及它们在转录本上的分布。从而推断出特定情况下哪些转录本正在被翻译。
多核糖体图谱:利用离心技术分离多核糖体和单一核糖体结合的mRNA,制备文库,在多核糖体部分检测到更高的丰度,可以推断单mRNA的翻译状态。
Ribo-seq:基于RNA足迹发展而来,使用环六胺抑制翻译伸长,导致核糖体在链上停止,然后用RNase1来消除mRNA,留下20多个受到核糖体保护的核苷酸,受保护的RNA测序揭示核糖体的密度和位置,用于产生文库。通过分析能够识别单个mRNA分子的相对翻译、起始、延伸、终止。
RNA-seq的其他应用
RNA在调节分子互作的生物学过程中发挥重要作用,RNA-seq能够用于解决分子内和分子间RNA的相互作用,揭示结构信息,深入了解转录和翻译过程。
互作分析的共同主题: enrichment of RNA that is interacting relative to RNA that is not.
RNA结构和RNA-蛋白质互作

1.结构化分析:利用核酸酶或试剂探测结构化RNA(双链、dsRNA)和非结构化RNA(单链、ssRNA),核酸酶降解后残余RNA被转化为cDNA并测序,读取深度和反应性呈正比。
PARS:RNA结构并行分析( parallel analysis of RNA structure)
2.化学作图:特异性修饰核糖核苷酸,标记阻断逆转录过程,导致cDNA截断,在修饰位点引起错掺突变,随后测序推断结构。
3.RRI分析法:RNA-RNA相互作用分析,首先将互作的RNA分子和生物素交联,然后进行富集,链接自由端,片段化之后制备文库,揭示相互作用的位点信息。
4.RNA-protein相互作用分析方法:RNA和蛋白质互作,交联免疫沉淀RNA后进行测序,利用UV紫外光辐射,在RNA和蛋白质间产生共价交联,片段化后,目标蛋白的抗体结合RNA,并链接适配器,提取cDNA,研究结构和互作体。
用分子内RNA互作探索RNA结构
查询RNA结构的方法有两种:核酸酶法和化学探测法
-
核酸酶法 如RNA结构并行分析、片段测序,使用特异酶降解后制备文库,结构化和非结构化区域通过计算识别,但其分辨率低于化学法,同时其尺寸比较大。
-
化学探测法 使用化学探针标记结构化或者非结构化区域,导致逆转录阻断或者cDNA误掺,测序揭示结构特征。探针是小分子,能够在体内确定,更具有生物学意义。还可以用于新生RNA分析和共转录RNA分析。
上述两种方法均产生短RNA片段,只报告单个位点,而误掺和突变检测法每次报告多个位点。所有方法都不是绝对有效,逆转录阻断法可能失败,化学标记可能突变,尖峰控制能提高检测质量却未普及。
这些方法都说明了RNA结构对基因和互作起到的重要作用。
分子间RNA的相互作用
全转录组法和靶向法类似,相互作用的RNAs在体内交联并富集,富集减少进入反应的RNA数量,起到调节作用。
RNA和蛋白质互作
芯片序列是理解RNA和蛋白质互作的关键工具,使用甲醛或者紫外线使RNA和蛋白质交联,再进行简单的RNA免疫沉淀测序得出结果。
该法的两个缺点:
- RNA和蛋白互作的温和洗涤条件下,许多非特异性结合片段也被富集。
- RNA断裂的缺失降低了结合位点的分辨率。
254nm紫外光交联剂介绍:能够在互作位点上产生共价键,并且不会交联蛋白质间互作,稳定了RNA和蛋白质的结合,允许严格富集,破坏天然RNA和蛋白质的互作,减少背景信号。
结论
作者预测RNA-seq将彻底改变真核转录组的分析方式,未来对于基因组的功能理解更加深入,各种分子调控至关重要,因此转录组发展并未结束。
目前,单细胞测序正成为许多实验室标准,空间转录组学也可能逐渐普及。长度测序方法可能取代二代测序技术,成为大多数人的默认方法,为了实现这种情况,需要提高吞吐量并降低错误率,如果长度测序能够像短读测序一样便宜可靠,那将会是未来首选技术!
文献:RNA sequencing: the teenage years,Nature Reviews Gentics,2019

本文由 mdnice 多平台发布