生信入门(六)——单细胞分析(Seurat)
生信入门(六)——单细胞分析(Seurat)
文章目录
- 生信入门(六)——单细胞分析(Seurat)
-
- 一、数据导入
-
- 1、数据来源
- 2、数据导入
- 二、标准预处理
-
- 1、QC和选择细胞进行进一步分析
- 2、规范化数据
- 3、识别高度可变的特征(特征选择)
- 4、缩放数据
- 三、主成分分析(PCA)
-
- 1、线性降维
- 2、确定数据集的“维度”
- 四、聚类细胞
- 五、非线性降维(UMAP/tSNE)
- 六、寻找差异表达的特征(簇生物标志物)
一、数据导入
1、数据来源
点击此处下载数据
数据说明:分析10X Genomics提供的外周血单细胞(PBMC)数据集,有2700个单细胞在illuminate NextSeq500上测序
2、数据导入
setwd("D:/RData/prac004")
# BiocManager::install("Seurat")
library(Seurat)
library(dplyr)
library(patchwork)
# 导入数据
pbmc.data<-Read10X(data.dir = "./filtered_gene_bc_matrices/hg19")
# 初始化seurat数据
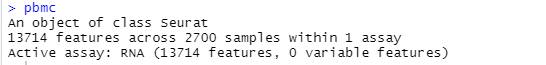
pbmc<-CreateSeuratObject(counts = pbmc.data,project = "pbmc3k",min.cells = 3,min.features = 200)
# pbmc
运行结果
二、标准预处理
该处包括基于QC指标(质控分析)、数据标准化和缩放以及高度可变特征检测的细胞选择和过滤
1、QC和选择细胞进行进一步分析
常用的QC指标包括:
-
在每个细胞中检测到的独特基因的数量。
低质量细胞或空液滴通常只有很少的基因 细胞双联体或多重联体可能表现出异常高的基因计数 -
同样,在细胞内检测到的分子总数(与独特的基因密切相关)
-
映射到线粒体基因组的读数百分比
低质量/垂死的细胞通常表现出广泛的线粒体污染 我们使用该PercentageFeatureSet()函数计算线粒体 QC 指标,该函 数计算源自一组特征的计数百分比 我们使用所有基因MT-的集合作为一组线粒体基因
#使用PercentageFeatureSet函数计算线粒体QC指标
pbmc[['percent.mt']]<-PercentageFeatureSet(pbmc,pattern = "^MT-")
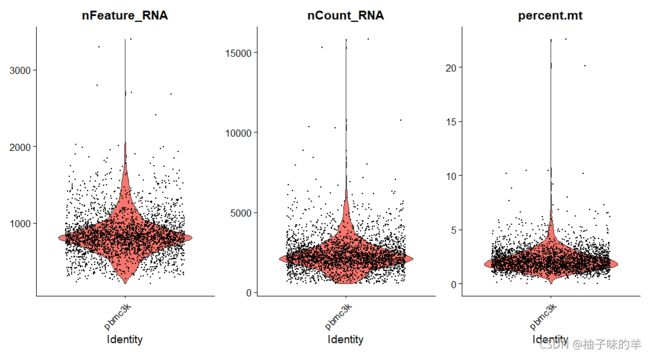
# 使用violin plot可视化 QC指标,并使用这些指标过滤单元格
VlnPlot(pbmc,features = c("nFeature_RNA","nCount_RNA","percent.mt"),ncol = 3)
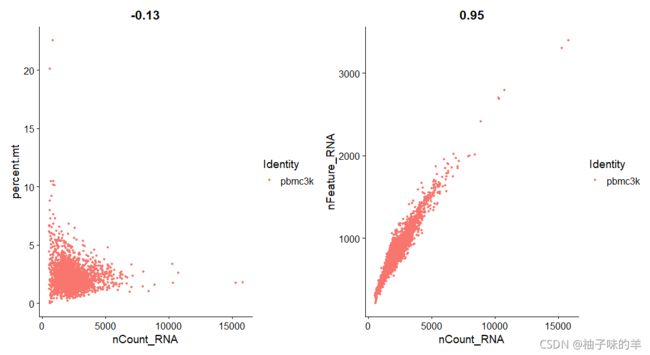
# FeatureScatter 通常用于可视化两个特征之间的关系
plot1<-FeatureScatter(pbmc,feature1 = "nCount_RNA",feature2 = "percent.mt")
plot2<-FeatureScatter(pbmc,feature1 = "nCount_RNA",feature2 = "nFeature_RNA")
plot1+plot2
# 将QC指标可视化,并使用这些指标过滤单元格
# 过滤具有2500或少于200的独特特征计数的单元格,过滤线粒体计数>5%的细胞
pbmc<-subset(pbmc,subset=nFeature_RNA>200 & nFeature_RNA<2500 & percent.mt<5)
运行结果
2、规范化数据
默认情况下,采用全局缩放归一化方法(LogNormalize),该方法将每个单元格的特征表达式测量值按总表达进行归一化,将其乘以比例因子(默认为10000),并对结果进行对数转换。标准化值存储在pbmc[[“RNA”]]@data中
# 数据规范化
pbmc<-NormalizeData(pbmc,normalization.method = "LogNormalize",scale.factor = 10000)
运行结果
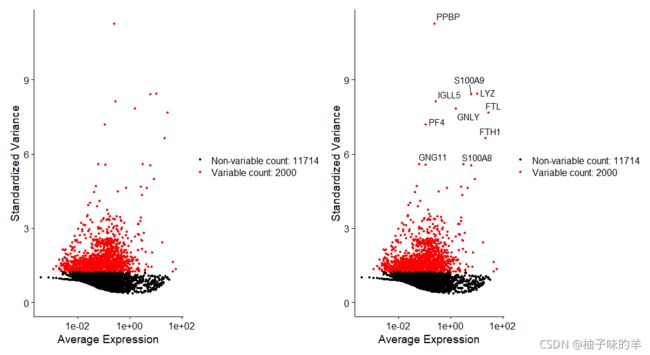
3、识别高度可变的特征(特征选择)
计算在数据集中表现出高细胞间变异的特征子集(即,他们在某些细胞中高度表达,而在其他细胞中表达低)——有研究发现,在下游分析中关注这些基因有助于突出单细胞数据集中的生物信号
# 识别高度可变的特征(特征选择)
pbmc<-FindVariableFeatures(pbmc,selection.method = "vst",nfeatures = 2000)
# 确定高表达的前十个基因
top10<-head(VariableFeatures(pbmc),10)
plot1<-VariableFeaturePlot(pbmc)
plot2<-LabelPoints(plot = plot1,points = top10,repel = TRUE)
plot1+plot2
运行结果
4、缩放数据
应用线性变换(“缩放”),这是在降维计数(如PCA)之前的标准预处理步骤。ScaleData()函数:
-
改变每个基因的表达,使跨细胞的平均表达为0
-
缩放每个基因的表达,使细胞间的方差为1
该步骤在下游分析中给予同等权重,因此高表达基因不会占主导地位 -
结果存储在pbmc[[“RNA”]]@scale.data
# 缩放数据
all.genes<-rownames(pbmc)
pbmc<-ScaleData(pbmc,features = all.genes)
运行结果

三、主成分分析(PCA)
1、线性降维
# 线性降维
pbmc<-RunPCA(pbmc,features = VariableFeatures(object=pbmc))
print(pbmc[["pca"]],dims = 1:5,nfeatures = 5)
运行结果

可视化处理
# Seurat提供可视化细胞和定义PCA,包括功能的几种有用的方法ViziDimReduction(),DimPlot()和DimHeatmap()
VizDimLoadings(pbmc,dims = 1:2,reduction = "pca")
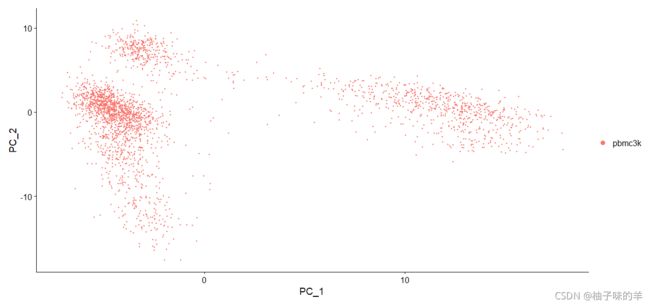
DimPlot(pbmc,reduction = "pca")
DimHeatmap(pbmc,dims = 1,cells = 500,balanced = TRUE)
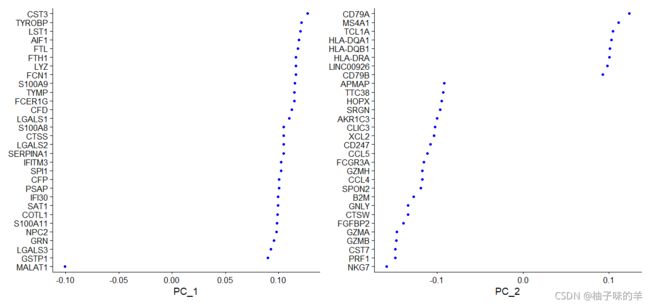

DimHeatmap()允许轻松探索数据集中异质性的主要来源,并且在尝试决定包含哪些PC以进行进一步下游分析时非常有用。单元格和特征都根据其PCA分数进行排序。设置cells为一个数字会在频谱的两端绘制极端单元格,这加快了大型数据集的绘制速度。
DimHeatmap(pbmc,dims = 1:15,cells=500,balanced = TRUE)
运行结果

2、确定数据集的“维度”
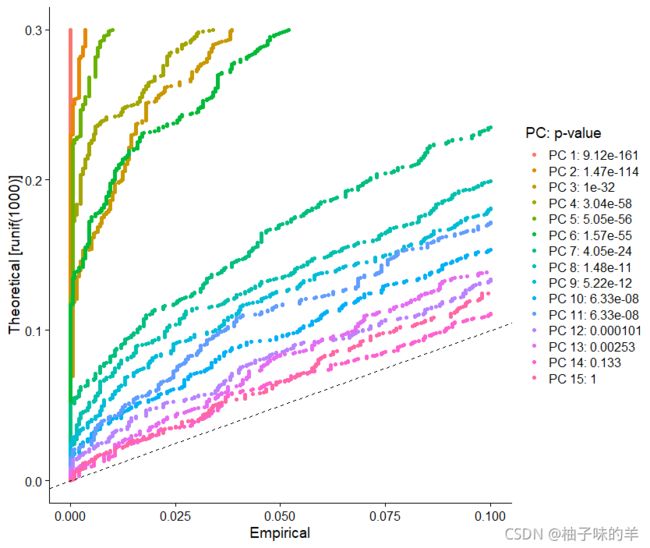
为了克服scRNA-seq数据的任何单个特征中的广泛技术噪音,Seurat根据其PCA分数对细胞进行聚类。每个PC基本代表一个“元特征”,该“元特征”组合了相关特征集的信息。因此,顶部主成分代表数据集的稳健压缩。但是,应该选择包含多少个组件?10?20?100?
# 确定数据集的维度
pbmc<-JackStraw(pbmc,num.replicate = 100)
pbmc<-ScoreJackStraw(pbmc,dims = 1:20)
运行结果
JackStrawPlot()函数提供了一个可视化工具,用于将每个PC的p值分布于均匀分布(虚线)进行比较。“显着”PC将显示出大量具有低p值得特征(虚线上方的实线)。在这种情况下,在前10-12个PC之后,重要性似乎急剧下降
# 可视化处理
JackStrawPlot(pbmc,dims = 1:15)
另一个启发式方法会生成一个“肘图”:根据每个(ElbowPlot()函数)解释的方差百分比对主成分进行排名。在这个例子中,我们可以观察到PC9-10周围的“肘部”,这表明大部分真是信号是在前10个PC中捕获的
ElbowPlot(pbmc)
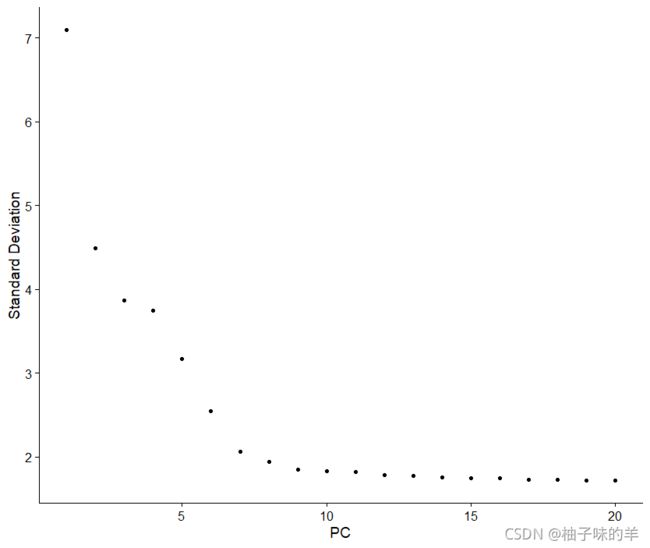
识别数据集的真实维度——对用户来说可能具有挑战性/不确定性。因此,我们建议考虑这三种方法。第一个是更受监督的,探索 PC 以确定异质性的相关来源,例如可以与 GSEA 结合使用。第二个实现了基于随机空模型的统计测试,但对于大型数据集很耗时,并且可能无法返回清晰的 PC 截止值。第三种是常用的启发式方法,可以即时计算。在这个例子中,所有三种方法都产生了相似的结果,但我们可能有理由选择 PC 7-12 之间的任何东西作为截止点。
我们在这里选择了 10 个,但鼓励用户考虑以下几点:
- 树突状细胞和 NK 爱好者可能会认识到与 PC 12 和 13 密切相关的基因定义了罕见的免疫亚群(即 MZB1 是浆细胞样 DC 的标志物)。然而,这些群体非常罕见,在没有先验知识的情况下,很难将这种规模的数据集与背景噪声区分开来。
- 鼓励用户使用不同数量的 PC(10、15 甚至 50 台!)重复下游分析。正如您将观察到的,结果通常不会有太大差异。
- 建议用户在选择此参数时将错误偏高。例如,仅使用 5 台 PC 执行下游分析会对结果产生显着的不利影响。
四、聚类细胞
# 聚类细胞
# 建立KNN图,并基于其局部领域中的共享重叠细化任意两个单元之间的边权重
pbmc<-FindNeighbors(pbmc,dims = 1:10)
#对细胞进行聚类,应用模块化优化技术,Louvain算法(默认)或SLM
# 以迭代方式将细胞分组在一起,目标是优化标准模块化函数
pbmc<-FindClusters(pbmc,resolution = 0.5)
head(Idents(pbmc),5)
运行结果

五、非线性降维(UMAP/tSNE)
Seurat提供了多种非线性降维技术,例如tSNE和UMAP,以可视化和探索这些数据集。这些算法的目标是学习数据的底层流形,以便将相似的单元格放在低维空间中。上面确定的基于图形的集群中的单元格应该共同定位在这些降维图中。作为UMAP和tSNE的输入,建议使用相同的PC作为聚类分析的输入
# 运行非线性降维(UMAP/tSNE)
pbmc<-RunUMAP(pbmc,dims = 1:10)
DimPlot(pbmc,reduction = "umap")

六、寻找差异表达的特征(簇生物标志物)
Seurat可以帮助找到通过差异表达定义聚类的标记。
# 寻找差异表达的特征(簇生物标志物)
# findmarkers为所有集群自动执行此过程,也可以测试集群组之间的对比,或针对所有单元格进行测试
# 默认情况下,ident.1与所有其他细胞相比,他识别单个簇的阳性和阴性标记。
# min.pct参数要求在两组细胞中的任何一组中以最小百分比检测到一个特征
# 而 thresh.test 参数要求一个特征在两组之间差异表达(平均)一定量
# 寻找cluster2的所有markers
cluster2.markers<-FindMarkers(pbmc,ident.1 = 2,min.pct = 0.25)
head(cluster2.markers,n=5)
# 寻找cluster5中与cluster0和cluster3n不同的所有markers
cluster5.markers<-FindMarkers(pbmc,ident.1 = 5,ident.2=c(0,3),min.pct = 0.25)
head(cluster5.markers,n=5)
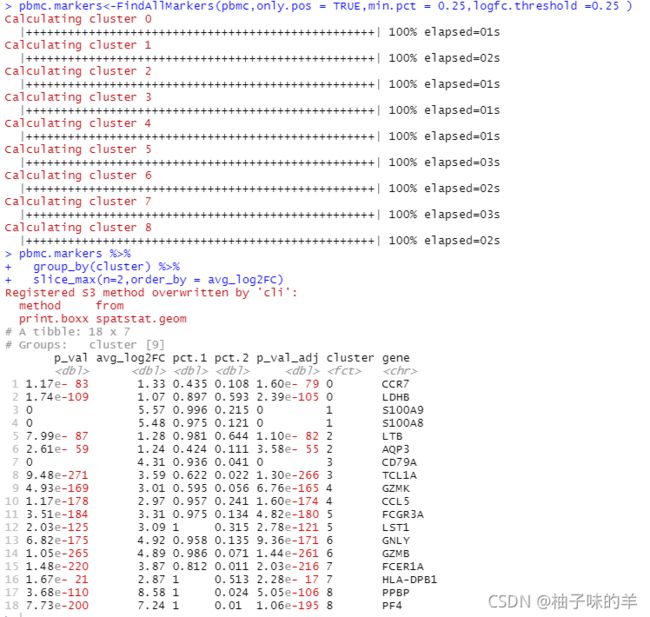
# 找出每个细胞簇的标记物,与所有剩余的细胞进行比较,只报告阳性细胞
pbmc.markers<-FindAllMarkers(pbmc,only.pos = TRUE,min.pct = 0.25,logfc.threshold =0.25 )
pbmc.markers %>%
group_by(cluster) %>%
slice_max(n=2,order_by = avg_log2FC)
运行结果

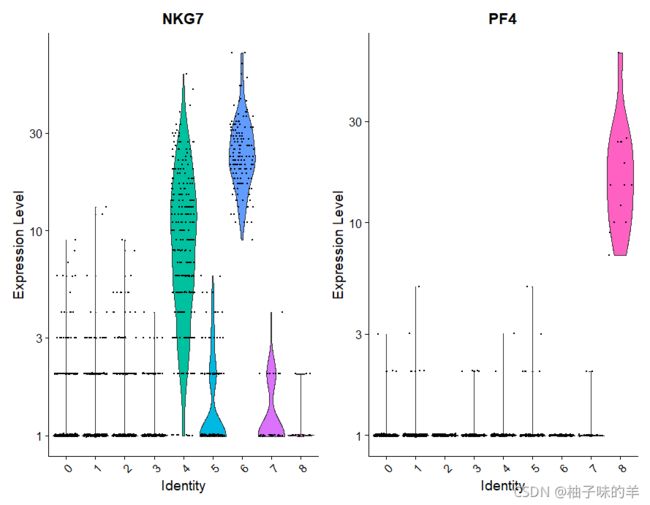
# 还有用于可视化标记表达的工具,Vlnplot(显示跨集群的表达概率分布)和FeaturePlot()(在tSNE或PCA图上可视化特征表达)是最常用的可视化,建议探索RidgePlot(),CellScatter(),和DotPlot()作为查看数据集的其他方法
VlnPlot(pbmc,features = c("MS4A1","CD79A"))
# 可以绘制行数据
VlnPlot(pbmc,features = c("NKG7","PF4"),slot = "counts",log = TRUE)
运行结果

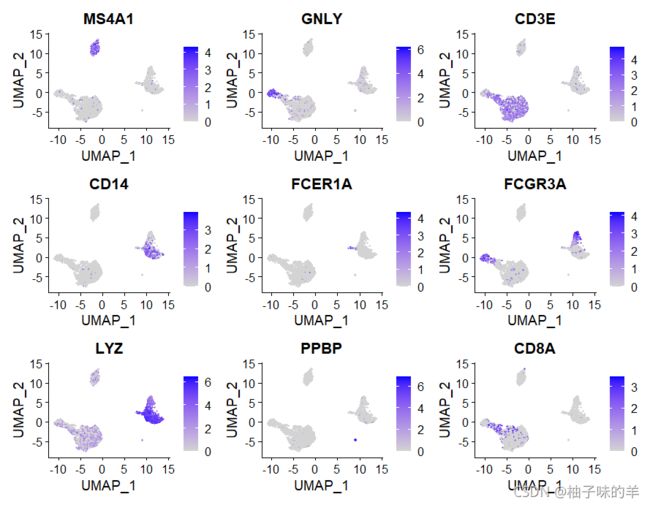
FeaturePlot(pbmc,features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
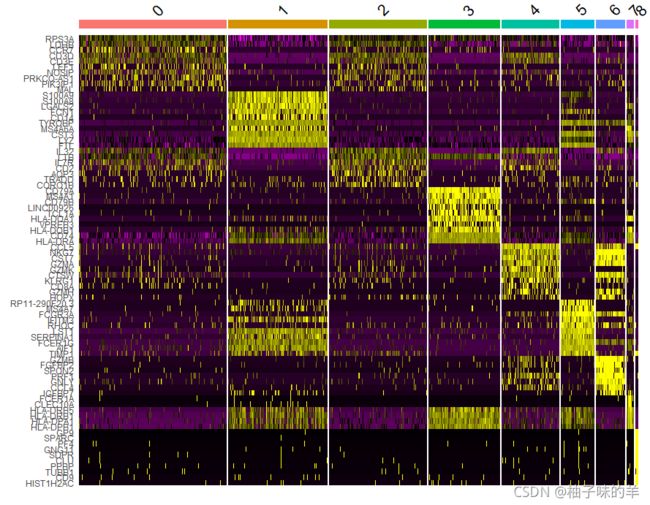
#DoHeatmap()为给定的细胞和特征生成一个表达热图。在这种情况下,我们为每个集群绘制前 20 个标记(或所有标记,如果小于 20)。
pbmc.markers%>%
group_by(cluster)%>%
top_n(n=10,wt=avg_log2FC)->top10
DoHeatmap(pbmc,features = top10$gene)+NoLegend()
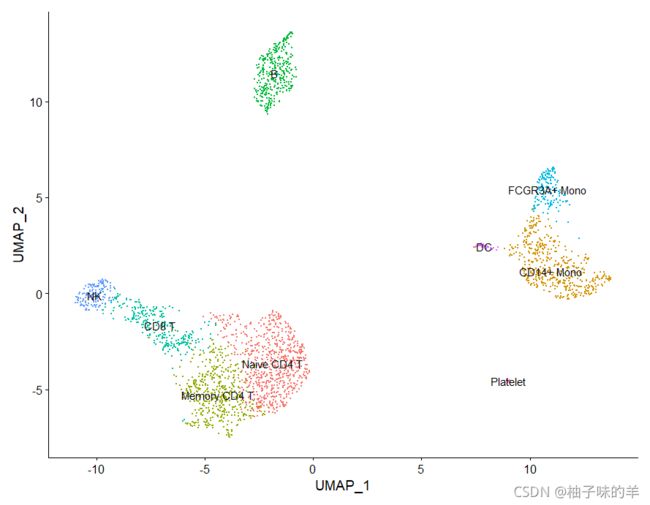
# 将细胞类型标识分配给集群
new.cluster.ids<-c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids)<-levels(pbmc)
pbmc<-RenameIdents(pbmc,new.cluster.ids)
DimPlot(pbmc,reduction = "umap",label = TRUE,pt.size = 0.5)+NoLegend()
saveRDS(pbmc,file = "./pbmc3k_final.rds")
告一段落