【语言模型生成分子更好】Language models can learn complex molecular distributions
Language models can learn complex molecular distributions【Nature Communications】
语言模型可以学习复杂的分子分布
paper : Language models can learn complex molecular distributions | Nature Communications
Data availability:
The processed data used in this study are available in GitHub - danielflamshep/genmoltasks: data, models and samples.
Code availability:
The code used to train models is publicly available.
JTVAE: GitHub - wengong-jin/icml18-jtnn: Junction Tree Variational Autoencoder for Molecular Graph Generation (ICML 2018).
CGVAE: GitHub - microsoft/constrained-graph-variational-autoencoder: Sample code for Constrained Graph Variational Autoencoders.
The RNN models were trained using the char-rnn code from GitHub - molecularsets/moses: Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models.
Trained models are available upon request.
使用的模型:
- 作者使用带有长期短期记忆的RNN模型,基于SMILES表示 (SM-RNN) 或 SELFIES表示 (SF-RNN)进行分子生成实验。
- 作者训练了两个最受欢迎的图生成模型:连接树变分自动编码器 (JTVAE)和约束图变分自动编码器 (CGVAE)进行对比。
评估的参数:
- 为了定量评估模型学习训练集分布的能力,作者计算生成分子的属性值和训练分子的属性值之间的Wasserstein距离。作者还计算了不同训练分子样本之间的Wasserstein距离,以确定最佳基准(TRAIN)。
- 作者使用了:药物相似性的定量估计 (QED)、合成可及性评分 (SA)、辛醇-水分配系数 (Log P)、精确分子量(MW)、Bertz复杂度 (BCT)、天然产物相似度(NP)等分子属性。
- 此外,作者还使用有效性(validity)、唯一性(uniqueness)、新颖性(novelty)等标准指标来评估模型的生成能力。
作者研究了语言模型学习复杂的分子分布的能力。通过编译更大、更复杂的分子分布,作者引入几个挑战性的分子生成任务评估语言模型的学习能力。结果表明,语言模型具有强大的生成能力,能够学习复杂的分子分布。语言模型可以准确生成:ZINC15数据集中惩罚 LogP得分最高分子的分布、PubChem数据集中多模态分子及最大分子的分布。
1.简介
其中语言模型循环神经网络 (RNN)以SMILES(Simplified Molecular Input Line Entry Specification)字符串的方式生成分子;其它一些模型或以图的方式生成分子(图生成模型),或将分子生成为3D空间中的点云。深度生成模型学习训练集的分布和生成有效的相似分子的能力对下游应用非常重要。
最初由于SMILES字符串表示的脆弱性,导致RNN模型经常生成无效分子。随后,研究人员使用鲁棒的分子字符串表示SELFIES(SELF-referencIng Embedded Strings),或者改进训练方法,RNN模型也能够持续生成高比例的有效分子。目前尚未有研究将语言模型用于生成更大更复杂的分子,或者从大规模化学空间进行生成。为了测试语言模型的生成能力,作者通过构建比标准的分子数据集更复杂的训练集来制定一系列挑战性的生成任务。作者在所有任务上训练语言模型,并和图生成模型进行比较。结果表明,语言模型的生成能力更强,比图生成模型能更好地学习复杂的分子分布。
2.结果
数据集以及定义了什么任务?
,表1 统计了与标准数据集Zinc和Moses相比,数据集中的原子数和环数(atom and ring number)。作者定义的三个任务(Log P, Multi, Large)都涉及具有更多子结构的更大分子,并且每个分子包含更大范围的原子数和环数。
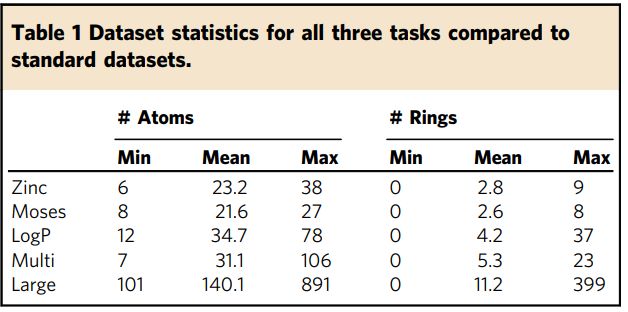 表1原子数和环数的统计信息
表1原子数和环数的统计信息
对于每项任务,作者通过绘制训练分子属性的分布以及语言模型和图生成模型学习的分布来评估生成模型的性能。作者对训练集和生成样本的分布使用直方图表示,并通过调整其带宽参数(所有模型使用一样的bandwidth )来拟合高斯核密度估计(KDE)。
所有模型生成1万个分子,计算它们的属性并生成所有绘图和指标。为了公平比较,在删除重复分子和包含在训练集中的分子后,作者使用相同数量的生成分子。
- 为了定量评估模型学习训练集分布的能力,作者计算生成分子的属性值和训练分子的属性值之间的Wasserstein距离。作者还计算了不同训练分子样本之间的Wasserstein距离,以确定最佳基准(TRAIN)。
- 作者使用了:药物相似性的定量估计 (QED)、合成可及性评分 (SA)、辛醇-水分配系数 (Log P)、精确分子量(MW)、Bertz复杂度 (BCT)、天然产物相似度(NP)等分子属性。
- 此外,作者还使用有效性(validity)、唯一性(uniqueness)、新颖性(novelty)等标准指标来评估模型的生成能力。
作者定义了三个生成任务:
(1)具有高惩罚LogP值的分子分布(图 1a,d)
(2)分子的多模态分布(图 1b,e)
(3)PubChem中最大的分子(图 1c,f)
Fig1 生成建模任务
a–c The molecular distributions defining the three complex molecular generative modeling task. 【a–c是在三项任务中分子分布图】
d–f examples of molecules from the training data in each of the generative modeling tasks.【d-f是三项任务中随机取的训练集样本】
- a The distribution of penalized LogP vs. SA score from the training data in the penalized logP task.【LogP 的分布规律,大多数位于4.0–4.5,10%超过6.0】
- b The four modes of differently weighted molecules in the training data of the multi-distribution task.
- c Large scale task’s molecular weight training distribution.
- d The penalized LogP task,
- e The multi-distribution task.
- f The large-scale task.
1、惩罚 LogP 任务(Penalized LogP Task.)
the penalized LogP task是为了搜寻化学空间,学会了分子分布,就具有高的penalized LogP,该任务目标是学习具有高惩罚LogP分数的分子的分布。作者在ZINC数据库中筛选惩罚LogP值超过 4.0 的分子构建训练数据集(160K个),它们的LogP得分大多数是4.0–4.5。在分子训练数据集的尾部(大概占总数据集的10%),它们有更高的LogP 值(大于6.0)。
For the first task, we consider one of the most widely used benchmark assessments for searching chemical space, the penalized LogP task—finding molecules with high LogP penalized by synthesizability and unrealistic rings.
We consider a generative modeling version of this task, where the goal is to learn distributions of molecules with high penalized LogP scores.
Finding individual molecules with good scores (above 3.0) is a standard challenge but learning to directly generate from this part of chemical space, so that every molecule produced by the model has high penalized LogP, adds another degree of difficulty. For this we build a training dataset by screening the ZINC15 database for molecules with values of penalized LogP exceeding 4.0. Many machine learning approaches can only find a handful of molecules in this range, for example JTVAE found 22 total during all their attempts. After screening, the top scoring molecules in ZINC amounted to roughly 160K (K is thousand) molecules for the training data in this task. Thus, the training distribution is extremely spiked with most density falling around 4.0–4.5 penalized LogP as seen in Fig. 1a with most training molecules resembling the examplesshown in Fig. 1d. However, some of the training molecules, around 10% have even higher penalized LogP scores—adding a subtle tail to the distribution.
结果如图2所示。语言模型表现更好,图2a中,SF-RNN产生的分布与训练分布更接近。CGVAE 和 JTVAE生成的大量分子的分数远低于训练集中最低分数。
在图2b-d 中,JTVAE和CGVAE能够生成比训练数据更大的 SA分数的分子,所有模型都能学习到LogP属性的主要分布,但RNN生成更接近的分布,QED属性同样如此。
The results of training all models are shown in Figs. 2 and 3. The language models perform better than the graph models, with the SELFIES RNN producing a slightly closer match to the training distribution in Fig. 2a. The CGVAE and JTVAE learn to produce a large number of molecules with penalized LogP scores that are substantially worse than the lowest training scores.
It is important to note, from the examples of these shown in Fig. 2a these lower scoring molecules are quite similar to the molecules from the main mode of the training distribution, this highlights the difficulty of learning this distribution.
In Fig. 2b–d we see that JTVAE and CGVAE learn to produce more molecules with larger SA scores than the training data, as well, we see that all models learn the main mode of LogP in the training data but the RNNs produce closer distributions– similar results can be seen for QED. These results carryover for quantitative metrics and both RNNs achieve lower Wasserstein distance metrics than the CGVAE and JTVAE (Table 2) with the SMILES RNN coming closest to the TRAIN oracle.
图2 惩罚LogP任务结果I
- a The plotted distribution of the penalized LogP scores of molecules from the training data (TRAIN) with the SM-RNN trained on SMILES, the SF-RNN trained on SELFIES and graph models: CGVAE and JTVAE. For the graph models we display molecules from the out of distribution mode at penalized LogP
as well as molecules with penalized LogP score in the the main mode [4.0,4.5] from all models.【训练数据和测试的四个模型它们的LogP分布,分布越接近说明越拟合的越好】
- b–d Distribution plots for all models and training data of molecular properties QED, LogP, and SA score.
表 2展示了(LogP, SA, QED, MW, BT, and NP这7种分子属性)Wasserstein距离 结果【其中TRAIN是一个oracle基础值,越接近它越好】,两个RNN生成的Wasserstein距离低于CGVAE和JTVAE,其中SM-RNN最接近最佳基准TRAIN。
Table 2 Wasserstein距离指标的结果
研究最高惩罚LogP区域(>= 6.0)
作者进一步研究了训练数据的最高惩罚LogP区域,即值超过6.0的训练分布的尾部。
- 图3e中,两个RNN都能学习到训练数据的这一区域,而图生成模型几乎完全忽略,只学习到更接近主模式的分子。
- 此外,训练数据中惩罚LogP得分最高的分子通常包含非常长的碳链和更少的环(图 3b,d),RNN能够学习到这一点。
- 这在模型产生的样本中非常明显,图 3f 显示的样本中,RNN主要产生具有长碳链的分子,而CGVAE和JTVAE产生的分子则具有许多环,语言模型学习的分布接近于图3a-d直方图中的训练分布。总体而言,语言模型比图模型可以更好地学习具有高惩罚LogP的分子分布。
We further investigate the highest penalized LogP region of the training data with values exceeding 6.0—the subtle tail of the training distribution. In the 2d distributions (Fig. 3e) it’s clear that both RNNs learn this subtle aspect of the training data while the graph models ignore it almost completely and only learnmolecules that are closer to the main mode. In particular, CGVAE learns molecules with larger SA score than the training data. Furthermore, the molecules with highest penalized LogP scores in the training data typically contain very long carbon chains and fewer rings (Fig. 3b, d)—the RNNs are capable of picking up on this. This is very apparent in the samples the model produce, a few are show in Fig. 3f, the RNNs produce mostly molecules with long carbon chains while the CGVAE and JTVAE generate molecules with many rings that have penalized LogP scores near 6.0. The language models learn a distribution that is close to the training distribution in the histograms of Fig. 3a–d. Overall, the language models could learn distributions of molecules with high penalized LogP scores, better than the graph models.
图3 惩罚LogP任务结果II
- a–d Histograms of penalized LogP, Atoms #, Ring # and length of largest carbon chain (all per molecule) from molecules generated by all models or from the training data that have penalized LogP ≥ 6.0.【生成的分子和训练数据集的LogP, Atoms #, Ring # and length of largest carbon chain】
- e 2d histograms of penalized LogP and SA score from molecules generated by the models or from training data that have penalized LogP ≥ 6.0.【生成的分子和训练数据集的LogP 和 SA得分】
- f A few molecules generated by all models or from the training data that have penalized LogP ≥ 6.0.【四种模型生成的和训练数据集中的少量分子图】
2、多分布任务(Multi-distribution task)
作者通过组合以下子集创建了一个数据集:
- (1) 分子量(molecular weight) (MW) ≤ 185的GDB13分子
- (2) 185 ≤ MW ≤ 425的ZINC分子
- (3) 哈佛清洁能源项目( CEP)分子,460 ≤ MW ≤ 600
- (4) POLYMERS分子,MW > 600。
多分布任务结果如图4所示,RNN模型能很好地捕获数据分布,并学习到训练分布中的每种模式(图4a)。JTVAE未能学习到GDB13的分布,对ZINC和CEP的学习也很差。同样,CGVAE学习到了GDB13的分布,但低估了ZINC的分布,未能学习到CEP的分布。
图4e中表明RNN模型能更紧密地学习训练分布,但CGVAE和JTVAE几乎没有区分主要模式。除CGVAE之外的所有模型都捕获了QED、SA和Bertz复杂度的训练分布(图4b-d)。
For the next task, we created a dataset by combining subsets of: (1) GDB13 molecules with molecular weight (MW) ≤ 185, (2) ZINC molecules with 185 ≤ MW ≤ 425, (3) Harvard clean energy project (CEP) molecules with 460 ≤ MW ≤ 600, and the (4) POLYMERS molecules with MW > 600. The training distribution has four modes– (Figs. 1b, e and 4a). CEP & GDB13 make up 1/3 and ZINC & POLYMERS take up 1/3 each of ∼200K training molecules.
In the multi-distribution task, both RNN models capture the data distribution quite well and learn every mode in the training distribution (Fig. 4a). On the other hand, JTVAE entirely misses the first mode from GDB13 then poorly learns ZINC and CEP. As well, CGVAE learns GDB13 but underestimates ZINC and entirely misses the mode from CEP. More evidence that the RNN models learn the training distribution more closely is apparent in Fig. 4e where CGVAE and JTVAE barely distinguish the main modes.
Additionally, the RNN models generate molecules better resembling the training data (Supplementary Table 4).
Despite this, all models– except CGVAE, capture the training distribution of QED, SA score and Bertz Complexity (Fig. 4b–d).
Lastly, in Table 2 the RNN trained on SMILES has the lowest Wasserstein metrics followed by the SELFIES RNN then JTVAE and CGVAE.
图4 多分布任务结果
Fig4 Multi-distribution Task:
- a The histogram and KDE of molecular weight of training molecules along with KDEs of molecular weight of molecules generated from all models. Three training molecules from each mode are shown.【在不同分子量(MW:Molecular Weight)的情况下,四个模型拟合train集的直方图】
- b–d The histogram and KDE of QED, LogP and SA scores of training molecules along with KDES of molecules generated from all models.【4中模型捕获QED, LogP and Bertz Complexity分布的情况】
- e 2d histograms of molecular weight and SA score of training molecules and molecules generated by all models.【在不同分子量(MW:Molecular Weight)的情况下,四个模型的SA得分,与train集更相似,说明RNN模型能更紧密地学习训练分布】
3、大规模任务(Large-scale task.)
①、该任务测试生成模型学习大分子的能力。
作者从PubChem中筛选了100多个重原子(heavy atoms),约有30万个分子,分子量(MW)1250 - 5000,大多数分子量在1250-2000之间(图1c)。
1、该任务中,CGVAE和 JTVAE都未能完成训练并且完全无法学习训练数据,所以Fig5a只有RNN而没有CGVAE和 JTVAE。对于非常长的分子字符串表示,SELFIES字符表示拥有额外的优势,SF-RNN可以更紧密地匹配数据分布(图5a)。使用SMILES语法生成有效的大分子更加困难,因为要为这些分子生成更多字符,并且模型出错并产生无效字符串的可能性更高,对比起来,SELFIES生成的从来不会invalid 。有趣的是,即使RNN模型生成的分子不在分布范围内并且比训练分子小很多,它们仍然具有类似的亚结构和与训练分子的相似之处(图5a)。
2、图5b显示训练分子具有较低和较高LogP值的模式(更少的环和更短的碳链生物分子定义为较低的模式,具有更多环和更长碳链的分子定义为较高的LogP模式),RNN模型能够学习训练数据分布的双模态性质。
In addition, the training molecules seemed to be divided into two modes of molecules with lower and higher LogP values (Fig. 5b): with biomolecules defining the lower mode and molecules with more rings and longer carbons chains defining the higher LogP mode (more example molecules can be seen in supplementary Fig. 8). The RNN models were both able to learn the bi-modal nature of the training distribution.
图5 大规模任务结果I
Fig. 5 Large-scale Task I:
- a The histogram and KDE of molecular weight of training molecules along with the KDEs of molecular weight of molecules generated from the RNNs. Two molecules generated by the RNN’s with lower molecular weight than the training molecules are shown on the left of the plot. In addition, two training molecules from the mode and tail of the distribution of molecular weight are displayed on the right.【大分子量的情况下,RNN可以很好地拟合训练分布】
- b The histogram and KDE of LogP of training molecules along with the KDEs of LogP of molecules generated from the RNNs. On either side of the plot, for each mode in the LogP distribution, we display a molecule from the training data.【训练分子和从RNNs产生的分子的较低和较高LogP值的模式的直方图和KDE(核密度估计)。在图的任一侧,对于LogP分布中的每个模式,我们显示来自训练数据的一个分子。】
②、作者针对训练数据包含各种不同的分子和子结构进行实验
图6a中,RNN模型充分学习到训练分子中出现的子结构的分布。即使训练分子变得越来越大并且出现的次数越来越少,两个RNN模型仍然能够生成这些分子(图5a,当分子量 >3000 时)。
The training data has a variety of different molecules and substructures, in Fig. 6a the RNN models adequately learn the distribution of substructures arising in the training molecules. Specifically the distribution for the number of: fragments, single atom fragments as well as single, fused-ring and amino acid fragments in each molecule. As the training molecules get larger and occur less, both RNN models still learn to generate these molecules (Fig. 5a when molecular weigh >3000).
PubChem 数据集中还包含很多肽(peptides)和环肽(cyclic peptides),作者对RNN模型生成的样本进行可视化分析,以评估它们是否能够保留主链结构(backbone chain structure)和天然氨基酸(natural amino acids)。我们发现,RNNs经常采样通常不相交的主链片段——被其他原子、键和结构打断。图6c展示了SM-RNN和SF-RNN生成的两个肽段示例。作者对RNN学习生物分子结构的能力进行了研究,图6b表明两个RNN模型都能学习必需氨基酸的分布。RNN模型也有可能用于设计环肽,作者展示了由RNN生成的分子,该分子与粘菌素和万古霉素具有最大的Tanimoto相似性(图6d)。
The dataset in this task contains a number of peptides and cyclic peptides that arise in PubChem, we visually analyze the samples from the RNNs to see if they are capable of preserving backbone chain structure and natural amino acids. We find that the RNNs often sample snippets of backbone chains which are usually disjoint—broken up with other atoms, bonds and structures. In addition, usually these chains have standard side chains from the main amino acid residues but other atypical side chains do arise.
In Fig. 6c we show two examples of peptides that are generated by the SM-RNN and SF-RNN. While there are many examples where both models do not preserve backbone and fantasize weird side-chains, it is very likely, that if trained entirely on relevant peptides the model could be used for peptide design. Even further, since these language models are not restricted to generating amino acid sequences that could be used to design any biochemical structure that mimic the structure of peptics or even replicate their biological behavior. This makes them very applicable to design modified peptides, other peptide mimetics and complex natural products. The only requirement would be for a domain expert to construct a training dataset for specific targets.
We conduct an additional study on how well the RNNs learned the biomolecular structures in the training data, in Fig. 6b we see both RNNs match the distribution of essential amino acid (found using a substructure search). Lastly, it is also likely that the RNNs could also be used to design cyclic peptides. To highlight the promise of language models for this task we display molecules generated by the RNNs with the largest Tanimoto similarity to colistin and vancomycin (Fig. 6d). The results in this task demonstrate that language models could be used to design more complex biomolecules.
图6 大规模任务结果II
Fig. 6 Large-scale Task II:
- a Histograms of fragment #, single atom fragment #, single ring fragment #, fused-ring fragment #, amino acid fragment # (all per molecule) from molecules generated by the RNN models or from the training data.【RNN生成的子结构和训练集的分布状况,越匹配说明拟合的越好】
- b Histograms of specific amino acid number in each molecule generated by the RNNs or from the training data.【由RNN生成的每个分子或训练集中特定氨基酸数量的直方图】
- c A peptide generated by the SM-RNN—MKLSTTGFAMGSLIVVEGT (right) and one generated by the SFRNN—ERFRAQLGDEGSKEFVEEA (left).【由两种RNN生成的肽取例】
- d Molecules generated by the SF-RNN and SM-RNN that are closest in Tanimoto similarity to colistin and vancomycin. The light gray shaded regions highlight differences from vancomycin.【展示了由SF-RNN和SM-RNN产生与粘菌素和万古霉素的Tanimoto相似性最接近的分子。浅灰色阴影区突出了与万古霉素的区别。这项任务的结果表明,语言模型可以用来设计更复杂的生物分子。】
③、作者还评估了所有模型生成分子的标准指标
(1)有效性:有效分子数与生成分子数之比,(2)唯一性:独特分子(非重复)与有效分子数之比,(3)新颖性:不在训练数据中的独特分子与独特分子总数的比率。每个任务生成1万个分子,结果如表3所示。JTVAE 和CGVAE具有更好的指标,具有非常高的有效性、唯一性和新颖性(均接近1),SM-RNN和 SF-RNN表现较差,但SF-RNN比较接近图生成模型。
We also evaluate models on standard metrics in the literature: validity, uniqueness and novelty. Using the same 10K molecules generated from each model for each task we compute the following statistics defined in ref. 17 and store them in Table 3: (1) validity: the ratio between the number of valid and generated molecules, (2) uniqueness: the ratio between the number of unique molecules (that are not duplicates) and valid molecules, (3) novelty: the ratio between unique molecules that are not in the training data and the total number of unique molecules. In the first two tasks (Table 3), JTVAE and CGVAE have better metrics with very high validity, uniqueness and novelty (all close to 1), here the SMILES and SELFIES RNN perform worse but the SELFIES RNN is close to their performance. The SMILES RNN has the worse metrics due to its poor grammar but is not substantially worse than the other models.
 表3 标准指标对比结果
表3 标准指标对比结果
3.讨论
在这项工作中,为了测试语言模型的能力,作者引入了三个复杂的分子生成任务,使用语言模型和图生成模型从具有挑战性的数据集中生成分子。结果表明,语言模型是非常强大、灵活的模型,可以学习各种不同的复杂分布,而图生成模型在很多方面表现较弱。
语言模型SM-RNN和SF-RNN在所有任务中都表现良好,优于基线方法。实验结果表明SF-RNN在每项任务中都有更好的标准指标,但SM-RNN有更好的Wasserstein距离指标。此外,SF-RNN比 SM-RNN具有更好的新颖性,这可能由于SELFIES语法导致语言模型只需记忆更少的训练数据。这也有助于解释为什么SF-RNN比SM-RNN具有更好的标准指标但更差的Wasserstein指标。此外,数据增强和随机SMILES可用于提高SM-RNN的新颖性得分。
图生成模型JTVAE和CGVAE不如语言模型灵活。对于惩罚LogP任务,得分为2的分子与得分为4的分子之间的差异很小。有时改变单个碳或其他原子会导致分数大幅下降,这可能解释了为什么CGVAE非常不适于该任务。对于多分布任务,JTVAE和 CGVAE表现较差的原因是:JTVAE必须学习广泛的树类型,其中许多没有类似环的较大子结构(GDB13分子),而另一些则完全是环(CEP和 POLYMERS);CGVAE必须学习大量不同的生成轨迹,这很困难,尤其是因为它在学习过程中只使用一个样本轨迹。出于同样的原因,这些模型无法训练 PubChem中大的分子。虽然语言模型可以灵活地生成更大的分子,但图生成模型更易于解释。
未来方向:对分子SMILES和SELFIES表示在深度生成模型中的使用进行更全面的评估;语言模型的改进,因为这类模型无法解释其他重要信息,如分子几何;探索语言模型在学习越来越大的化学空间方面的能力。