Pssp-mvirt: 基于多视图深度学习架构的肽二级结构预测
目录
摘要
介绍
方法和材料
数据
初始数据集集合
训练和测试数据集
建议的 pssp-mvirt 的架构
多视点特征融合模块
功能视图1-顺序信息嵌入
功能视图2-进化信息嵌入
功能视图3-隐藏状态信息嵌入
多视点特征融合策略
高隐含特征提取模块
基于新填充技术的CNN局部特征提取
基于 bgru 的全局特征提取
门控复发单位
附加限制机制
特征表达能力增强模块
多头注意机构
预测模块
绩效指标
实验环境
结果和讨论
与现有二级结构预测方法的比较
多肽二级结构预测的长度偏好研究
确定模型的最优网络体系结构
我们的多视角特征融合策略的影响
个案研究
网站
结论
摘要
肽二级结构的预测对揭示肽的作用机制具有重要意义,具有潜在的应用前景。本文提出了一种基于多视点信息、限制和迁移学习的多视点深度学习方法——肽二级结构预测法。为了充分利用鉴别信息,提出了一种多视点融合策略,分别从序列信息、进化信息和隐藏状态信息等多个视点整合不同的信息,生成统一的特征空间。此外,我们构建了一个混合网络结构的卷积神经网络和双向门控循环单元提取全局和局部特征的多肽。此外,我们利用迁移学习有效地缓解了训练样本(具有实验验证结构的多肽)的缺乏。独立测试的比较结果表明,我们提出的方法明显优于最先进的方法。特别是,我们的方法在分段级别表现出更好的性能,这表明我们的模型在捕获局部区分性信息方面有很强的能力。实例研究还表明,我们的 pssp-mvirt 在预测新的肽二级结构方面取得了良好的效果。重要的是,我们建立了一个 web 服务器来实现这个建议的方法,目前可以通过 http://server.malab.cn/pssp- mvirt 访问这个方法。我们希望它能成为研究人员感兴趣的有用工具,促进我们的方法的广泛应用。
介绍
肽具有高特异性、高耐受性、高渗透性、副作用少、生产成本低、易于制造和修饰等优点,近年来已成为治疗各种疾病的潜在治疗分子[1]。多肽的生物学功能与其结构密切相关。因此,了解生物活性肽的结构不仅有助于进一步了解肽的功能,而且还可以指导设计具有所需功能的肽[2]。二级结构是指蛋白质大分子的三维局部片段,这些片段是在氨基酸残基连接成一个序列之后,在蛋白质折叠成三级结构之前形成的。二级结构包括沿着骨架的氢键,使长链折叠成局部形状,主要是螺旋(h)、股(e)和线圈(c)[3]。随后,在预测肽的三级结构之前,一个重要的步骤是确定肽的二级结构,这可以提供关于结合特性和骨架的信息,有助于三级结构的预测。
在过去的几年里,已经提出了几种基于机器学习的预测蛋白质二级结构的计算方法。例如,Jones[4]设计了一个两阶段神经网络,其进化特征来源于特定位置记分矩阵(PSSM),这是一种包含足够进化保守信息的轮廓。周和特罗扬斯卡亚[5]提出了一种新的基于监督生成随机网络的方法,该方法从条件分布中学习马尔可夫链,并将其应用于蛋白质结构预测。后来,王等人又提出了自己的观点。[6]提出了一种深度卷积神经场模型,该模型不仅可以探索复杂的序列-结构关系,而且可以探索相邻属性之间的相互依赖关系。特别是,与以往的其他方法不同,它可以在没有紧密同源关系或几乎没有进化信息的情况下为蛋白质提供更准确的二级结构预测。类似地,Li和Yu[7]提出了扩散卷积递归神经网络(DCRNN),这是一种端到端的深度神经网络,它注重利用全局特征和局部特征,并利用多任务学习来同时预测二级结构和氨基酸溶剂可及性。为了捕捉与蛋白质一起的长距离依赖,Heffernan等人。[8]设计了一种具有长短期记忆机制的双向递归神经网络Spider3,旨在提取全局特征。他们证明,Spider3的性能优于其他以前的方法。Busia和Jaitly[9]提出了NextStep条件深卷积神经网络(CNN),它通过使用一种新的链式预测方法对现有技术进行了改进。神经网络将二级结构预测框定为下一步预测问题。最近,方等人。[10]开发了一个深度起始内部网络(即Deep3I),该网络集成了各种信息,如氨基酸的物理化学性质,以及来自PSI-BLAST轮廓(PSSM)的进化信息,以训练预测模型。Deep3I能够有效地处理每个残基之间的局部和全球相互作用,从而做出准确的预测。除了上面介绍的方法外,还有其他优秀的蛋白质二级结构预测方法,如PSIPRED[11]、JPRED[12]、RaptorX[13]、PHD[14]、PROTEUS2∗[15]等。
然而,上述方法都是专门为蛋白质二级结构预测而设计的,蛋白质和多肽的二级结构有很多不同之处。一方面,以往的研究已经证明,通过比较多肽和蛋白质的二级结构组成,对于蛋白质和多肽中一些相同的残基片段,它们的二级结构是不同的[2]。另一方面,缺乏准确的肽二级结构也限制了对肽功能的预测,如抗癌活性[16],这严重依赖于序列信息。因此,通过整合额外的二级结构信息来期望改进是合理的。为了解决这个问题,Singh et al.。[2]首次提出了一种基于随机森林的方法PEP2D,该方法利用序列信息和进化信息来预测多肽的二级结构,并通过利用二级结构信息进行了大量的改进。综上所述,多肽的二级结构预测对于下游结构或功能的预测具有重要意义。
在这项研究中,我们提出了一种新的深度学习神经网络,称为基于多视图信息的肽二级结构预测,约束和转移学习(PSSP-MVIRT),它是专门为肽二级结构预测设计的。提出的PSSP-MVIRT的新颖性可以归结为以下三个方面。首先,为了充分利用判别性信息,我们采用了多视角融合策略,分别从序列信息、进化信息和隐藏状态信息等多个角度对信息进行融合。其次,为了提取多肽的全局和局部特征,我们使用了CNN[28]和双向门控递归单元(BGRU)的混合网络结构。特别是,我们引入了一种额外的约束机制,可以捕获高潜在特征表示,提高表示能力。第三,由于缺乏具有实验验证结构的训练样本,本文首先利用迁移学习在大规模蛋白质数据集上训练我们的模型,然后对模型进行微调以进行肽二级结构预测。在基准数据集上的大量对比实验表明,我们提出的方法在独立测试上的性能明显优于最先进的方法。更重要的是,通过对比分析,我们的方法能够捕捉到更多的多肽的局部信息特征,这可以有效地帮助提高预测性能。
方法和材料
数据
初始数据集集合
在这项研究中,我们使用了相同的基准数据集,即Scratch-1D,这是几个研究中通常用于绩效评估的数据集[17]。该数据集由5772个蛋白质数据的一级结构和相应的二级结构组成,具有三种结构状态(H、E和C)。在Scratch-1D中,蛋白质结构是通过X射线晶体学得出的至少2.5埃的分辨率,没有断链,少于5个未知氨基酸,长度至少30个残基。值得注意的是,数据集中的序列一致性被降低到25%,以避免性能评估的偏差。然而,我们发现有一些蛋白质含有用符号X表示的非自然残基。去除这些多肽后,我们的数据集中保留了4542个蛋白质和多肽序列。
训练和测试数据集
由于我们的任务是预测肽的二级结构,其样本通常小于100个残基长度,数据集中长度大于100个残基长度的蛋白质序列被分割为100个残基长度,而不是使用全长的蛋白质序列。通过这样做,我们总共得到了9262个分段蛋白质子序列。这样做的原因是为了更好地捕捉短肽样序列的特征,以获得更好的性能。所有分段蛋白质子序列用于预训练初始的基于深度学习的预测模型,肽序列用于模型的微调以生成任务特定的模型。对于肽模型训练阶段,我们从1285个肽序列中随机选取1028个作为训练数据集,其中 h、 e 和 c 三个结构状态的数目分别为38749、18020和32910(表1)。其余257个肽序列(h 为7450,e 为4199,c 为6957)作为我们的测试集,用于模型性能评价。每个肽的序列长度在30到100个残基之间,这些残基用三态二级结构标记。三态二级结构和氨基酸测序的统计数据见图1 a 和 c,其中每种颜色在每个 fasta 文件中的长度分别表示螺旋(h)、链(e)或线圈(c)的数量。图1b 显示了数据集中每个状态的指定序列对应的肽序列的数量。在这项工作中使用的数据集的详细信息可以在表1中看到。图1还说明了数据集的统计。

表1. 这项工作中使用的数据集的摘要

图1。肽数据集统计。(a)每个残基肽序列中每个二级结构的数目 x; (b)相应的残基肽序列数目 x 与特定的羟乙基胆碱酯酶含量相关; (c)每个无残基肽序列中每个二级结构的数目 x; (d)相应的无残基肽序列数目 x 与特定的羟乙基胆碱酯酶含量相关。
建议的 pssp-mvirt 的架构
图2说明了所建议的神经网络的结构,即 pssp-mvirt。该方法包括四个主要模块: (1)多视点特征嵌入、(2)特征提取、(3)特征表示能力增强和(4)预测模块。预测过程描述如下。在模块(1)中,给定一个氨基酸测序,它首先被编码成三个特征度量,分别代表序列信息、进化信息和隐藏状态信息。然后,为了学习一个统一的特征嵌入,我们使用基于多视图融合策略的余弦距离来衡量两个嵌入特征的相似程度。在模块(2)中,为了进一步挖掘更多的鉴别信息,我们使用了 cnn 和 bgru 的混合神经网络,捕捉局部特征和全局特征。在模块(3)中,我们使用了广泛使用的自然语言处理技术——Transformer Encode[18] ,以增强从最后一步得到的特征表示。最后,在模块(4)中,结果特征被输入到我们的模型中,以预测肽属于哪个结构状态: c,h 或 e 的每个位置,下面详细介绍了这四个模块

图2.PSSP-MVIRT的体系结构。(A)用4种特征表示方法对多肽进行编码,以探索不同的序列信息,然后通过级联(B)CNN和填充技术提取局部特征和并行BGRU在分段水平提取局部-全局特征来整合特征矩阵;(C)通过多头注意机制增强所得到的特征;(D)通过训练良好的模型预测多肽的二级结构,并利用专门用于二级结构可视化的工具PyMol进行可视化。
多视点特征融合模块
在这一部分中,我们介绍了如何将我们的原始多肽序列预处理成数字特征表示,这些特征表示可以用机器学习算法进行训练。下面,我们首先从进化信息、序列信息、隐藏状态信息和相似性信息四个特征视图来介绍嵌入方法。其次,为了生成统一的特征空间,我们采用了多视角的特征融合和学习策略。
功能视图1-顺序信息嵌入
这里的顺序信息由word2vec[19]从索引列表中生成。与一热编码序列信息相比,它可以学习具有潜在语义的高质量残基向量,并防止零冗余表达。
功能视图2-进化信息嵌入
PSSM是一个m∗n矩阵,其中m是每个蛋白质序列的长度,n是标准残基的数目。PSSM分数通常显示为正整数或负整数。通过这种方式,我们可以计算出序列中特定位置的20个氨基酸的特定位置分数。得分较低的氨基酸有很大的趋势进化为得分较高的氨基酸,保持稳定状态。在这项研究中,每个肽序列的PSSM是通过使用默认参数对SwissProt数据库[21](版本于2020年9月5日更新)进行三次位置特定迭代基本局部比对搜索工具(PSI-BLAST)+[20]来生成的。
功能视图3-隐藏状态信息嵌入
隐马尔可夫模型(HMM)是随机模型的一种。该方法被广泛应用于蛋白质二级结构的预测。在多肽二级结构预测中,通过隐马尔可夫模型学习H(螺旋)、E(链)和C(螺旋)等结构,并将这些隐马尔可夫模型应用于二级结构未知的新的肽序列。来自HMM的概率的输出被用于预测序列的二级结构[22]。在本研究中,显性序列是多肽序列,隐藏状态是它们的二级结构。我们在研究中使用的HMM配置文件是从HMMER3.0[23]生成的
多视点特征融合策略
通过融合上述三个特征视图的信息,我们使用余弦相似度来生成统一的特征表示空间。给定具有n个序列的基准数据集{P,E},其中P表示PSSM,E表示嵌入的顺序信息。对于每个给定肽,其特征可以表示为矩阵X,如下所示:

为了简化积分过程,将矩阵1范数替换为无穷范数,如下所示。此外,我们还集成了一些补充信息,它由两部分组成:(1)由PSSM信息和嵌入序列信息产生的相似性信息;(2)由HMM信息和嵌入序列信息产生的相似性信息。隐马尔可夫模型(HMM)和嵌入序列信息的统一特征表示空间的生成过程同上。

其中?p?∞是矩阵P的无穷范数,?h?∞是矩阵H的无穷范数。最后,HMM、PSSM和两个统一的特征表示空间连接成一个m×w矩阵作为高潜在输入特征,其中m是肽长度的长度,w是HMM、PSSM和两个补充信息的宽度之和。
高隐含特征提取模块
对于高潜在特征提取,我们采用了CNN和BGRU的混合神经网络,其中CNN用于提取局部特征,而BGRU用于提取全局特征。
基于新填充技术的CNN局部特征提取
在这里,我们利用CNN来学习和提取局部特征。每个卷积神经元只处理其感受野的数据。因此,本文使用CNN来提取多肽特征表示中的局部信息。值得注意的是,在每个二维卷积层之前使用了支持信息中讨论的填充技术(循环填充和反射填充),如图3所示。通过使用填充技术,我们可以有效地解决肽链的边界信息提取问题,提高了对每个肽链末端的预测性能。在补充材料中介绍了填充技术的更多细节。

图3.特征提取模块中的局部特征提取部分。
基于 bgru 的全局特征提取
通过附加约束,将全局特征提取分为两部分。在第一个全局特征提取部分,使用完全连接的层作为过渡层局部特征提取部分和全局特征提取部分之间的关系。然后,bgru 接收到一个更有效的特征矩阵,进一步提取远程依赖关系。在第二个全局特征提取部分,将完全连通层插入到BGRU层的后面,如图2所示。本文还探讨了将多肽分成多个子序列作为 bgrus 的输入是否具有更好的性能,并将其命名为并行 bgru,其结构见图4。在“我们模型的最佳网络结构的确定”一节中讨论了不同级别的并行 bgru 结构的对比实验。

图4,并行 bgru 的架构
门控复发单位
Gru [24]在解决标准递归神经网络的消失梯度问题(rnn)方面有很好的表现。Gru 允许每个循环单元自适应地捕获不同时间尺度的依赖性,如图5所示。对于每个单元来说,使用 grus 来记忆在一系列长时间的时间步骤中输入流中存在的特定特性是更容易的。

其中 zt 被定义为一个更新门,控制前一个时间的状态信息进入当前状态的程度; rt 被定义为一个复位门,它控制如何复位许多信息被写入候选激活∼ ht 从以前的状态。

附加限制机制
在模型中引入了附加约束机制。如图6所示,当肽序列的二级结构相同时,如果它接收到不同的肽序列,则它在神经网络的中间应该具有相似的表示。为此,在全局特征提取部分之间插入了附加的约束机制。附加限制由用于重塑瞬变状态的完全连接的层组成。它接收第一全局特征提取部分的输出作为输入特征。在完全连接层之后,使用二级结构标签和完全连接层的输出,通过均方误差作为成本函数来计算额外的损耗,如图7所示。

图6.神经网络中间的特征表示与预测结果之间的关系示例。

图7.PSSP-MVIRT中的附加限制机制
特征表达能力增强模块
该部分主要由the six-stacked eight-head Transformer Encoder[18]组成。特征提取部分的输出被接收为嵌入的输入,这可能是一种比单词嵌入更有效的表示方式。它通过将这些向量馈送到自注意力层,然后馈入前馈神经网络来处理高层特征,最后将输出发送到下一个变压器编码块。经过变压器编码器的处理后,得到两个完全相连的层,这两个层接收注意特征并输出二级结构标签。
多头注意机构
“注意”的概念最近在神经网络训练中得到了广泛的应用,特别是在翻译和对齐单词方面,这类似于肽的二级结构预测,因为它可以灵活地捕捉全局和局部依赖。在模型设计中,我们尽可能紧跟原有的变压器编码器部分,作为主要的特征增强部分。
结果表明,将查询、关键字和值分别线性投影到DK、DK和DV维度的线性投影次数h次,优于使用dModel维关键字、值和查询的单一注意函数。每个变压器编码块都包括一个按比例扩展的点积关注层和带有剩余连接机制的完全连接。整体的多头注意机制如下

是其中Q表示查询矩阵;K表示关键字矩阵;V表示值矩阵;WQ、WK、WV分别表示训练权重矩阵。
预测模块
为了训练一个稳健的预测模型,我们构造了一个新的损失函数,它由以下两个代价函数组成:(1)均方误差中约束函数和(2)加权均方误差损失函数,如下所示。为了平衡这两个成本函数,使用平衡系数γ来计算优化器的最终成本,如下所示:
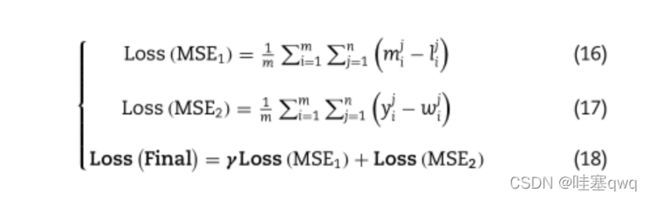
其中MSE是均方误差的首字母缩写,m定义为附加限制部分的输出,l定义为一热编码器编码的二级结构标签,y定义为特征提取模块的输出,w定义为加权编码标签,状态E的权重为1.25,其余为1,m为样本数,n为无零填充的多肽样本长度。
绩效指标
在这项研究中,PSSP-MVIRT的性能通过每个结构状态Acci(AccH,ACCE,ACCC)的预测精度,所有状态即Acc的预测精度,每个结构状态的精度和片段重叠度量[26](SOV)来衡量。这些指标的计算方法如下:

其中i是任意二级结构元素(螺旋、片状或卷曲);Ai是每个状态中正确预测残基的总数;Aii是状态i中正确预测残基的数量;αi是状态i在整个测试集中的比例;S1和S2是与实际和预测的二级结构相对应的片段;len(S1)对应于定义片段S1的残基的数量;minOv(S1,S2)对应于重叠的S1和S2片段的长度;max ov(S1,S2)是其中任何一个片段在状态I具有残基的S1和S2片段的最大重叠;δ(S1,S2)计算如下

实验环境
为了获得更好的性能和加快网络的训练速度,在PSSP-MVIRT中采用了批归一化和丢弃技术。在除附加限制部分之外的每一层之间插入忽略速率p为0.25的丢弃层。对于批量归一化,它被插入在(1)输入和特征融合部分,(2)特征融合部分和局部特征提取部分,(3)局部特征提取部分和第一全局特征提取部分,(4)第一全局特征提取部分和第二全局特征提取部分,以及(5)第一全局特征提取部分和附加约束部分之间。它被认为具有通过增加控制层输入的均值和方差的网络层来减少内部协变量变化的能力[27]。
当涉及到附加成本函数时,二级结构标签由一热编码器编码,平衡系数γ被设置为0.1.。在PSSP-MVIRT神经网络中,所有卷积层、BGRU层和部分全连通层均采用修正线性单元(RELU)激活函数。在附加限制部分中,在最终完全连接层之前使用激活函数Sigmoid,并且在第二全局特征提取部分中,在最终完全连接层之前使用激活函数Softmax。
我们的深度学习模型共有31744430个参数,通过ADAM算法进行全局训练,学习速率l=1e−4,以最小化代价函数损失(最终)。训练epoch设置为250,在97表现最好(附图S4)。所有的训练和测试过程都是基于NVIDIA Titan RTX GPU执行的,并使用基于PyTorch的PYTHON实现。
结果和讨论
与现有二级结构预测方法的比较
为了评估我们提出的PSSP-MIRVT的有效性,我们在相同的独立测试集上将其与现有流行的蛋白质二级结构预测方法如PHD[16]和Jpred[14]进行了比较。值得注意的是,对于我们的预测方法,我们训练了三个不同的加权模型,以不同的结构状态权重来避免数据不平衡的问题。评价结果如表2所示。在五个模型中,我们的PSSP-MIRVT的不同加权预测模型表现良好,其中E状态权重为1.25的模型性能最好,分别为ACC 78.50%、AccH 90.16%、ACCE 56.84%、ACCC 68.47%和SOV 75.81%。我们观察到我们的多肽专一性方法明显优于蛋白质设计的方法,特别是在多肽二级预测中的SOV,这表明为蛋白质二级结构预测而设计的方法不能充分捕捉了短肽序列的区分性信息,证明了针对多肽设计的PSSP-MIRVT的必要性。与PHD相比,我们的模型在几乎所有指标上都优于PHD,在ACC、AccH、ACCE和SOV方面分别获得了2.19、9.28、6.26和17.92%的性能提升。与Jpred相比,我们的模型在几乎所有指标上都比Jpred高0.45、10.92、4.30和15.19%的ACC、AccH、ACCE和SOV。可以看出,与Jpred和PHD具有较好的ACC但较差的AccH和ACCE不同,我们的模型不仅获得了具有竞争力的ACC、ACCE和ACCC,而且达到了相当好的AccH,比现有方法高出10%以上,证明了PSSP-MIRVT在处理标签不平衡困难方面的优势。此外,我们使用SOV,这是在细分市场级别提供测量的另一个重要指标,来评估方法的整体性能。如表2所示,我们的PSSP-MIRVT在SOV上可以获得非常出色的性能,比以前的方法超过15%。我们推测,特征融合后的卷积层使我们的模型能够更好地捕捉多肽局部区域的信息。因此,它在多肽片段水平上表现出比现有方法更好的性能。值得注意的是,我们的方法是一种端到端的深度学习方法,只需从序列中学习和提取特征并进行预测,而不像传统的基于机器学习的方法那样进行任何专业的特征工程。综上所述,我们的模型(E-State权重为1.25)在预测多肽二级结构方面比Jpred和PHD更有效,尤其是对AccH和Sov。

多肽二级结构预测的长度偏好研究
为了进一步研究我们的模型是否对肽二级结构预测具有长度偏好,我们将测试集分为四个不同长度间隔的子集:[30,35),[35,40)、[40,45]和(45,50]个残基。四个子集的详细情况可在补充表S2中找到。PSSP-MIRVT在四个测试子集上进行评估,结果显示在补充表S1中。图8描述了我们的方法在[30,35),[35,40),[40,45]比(45,50)区间的性能更好,最高的ACC分别为75.10,78.93和81.58%,SOV分别为64.58,78.30和77.53%。相比之下,我们在[30,35]的ACC和SOV分别领先1.41%和8.31%,在(35,40]的ACC和SOV领先3.3和22.52%,在(40,45)的ACC和SOV领先1.26%和20.80%。有趣的是,我们发现随着多肽长度的增加,性能呈现出明显的下降趋势(图8),这表明我们的方法可以达到预测较短多肽的最佳性能。这表明,我们的模型在预测长度小于30个残基的多肽方面具有优势,而现有的方法并不擅长。此外,我们还在不同长度区间的三个子集上对现有的其他方法进行了评估,结果显示在补充表S2中。不幸的是,在我们的模型中没有观察到明显的趋势。

确定模型的最优网络体系结构
为了确定模型的最优网络结构并获得最佳性能,我们对模型的两个主要超参数进行了优化,其中一个是卷积层数,另一个是所提出的BGRU的段数。对于最佳卷积层数的确定,我们分析了从1到4的不同层数。结果如附图S5A所示。结果表明,我们的模型达到了最大值,当层数达到3时,ACC值为78.50%,ACCH值为90.16%,ACCE值为56.84%,ACCC值为68.47%。具体地说,与单层卷积层相比,具有三层卷积层的模型的ACC和SOV分别提高了0.93和1.07%,表明我们可以用三层卷积来捕捉最充分的信息。一个或两个卷积层性能较差的原因可能是缺乏局部特征信息提取,而四个2D卷积层性能较差的原因可能是训练参数过多,导致过拟合。
同样,为了优化所提出的并行 bgru 体系结构,我们调查了不同的段数,范围从1到4,并在补充图 s5b 中说明了结果。值得注意的是,如果片段数设置为1,它就是没有任何片段的原始肽。正如预期的那样,在段数等于1的情况下,我们取得了最好的性能,因为分割会导致全局信息的丢失。具体来说,最佳ACC和SOV分别为78.50% 和75.81% ,分别比第二最佳结构模型高0.94% 和2.59% 。此外,我们的1、2、3段模型表现良好,证明在肽段水平上提取的特征可能是一种新的探索方法。然而,如果多肽被分割成太多的子序列,比如4个或更多的片段,如图 s5b 所示,性能会显著下降,其潜在原因可能是局部结构模式被打破。此外,我们还研究了我们的模型的学习率的影响。详细的结果可以在补充材料中找到。
我们的多视角特征融合策略的影响
为了分析多视点特征融合策略的影响,我们将融合后的特征分别与PSSM Profile、HMM Profile和word2vec中提取的三个独立特征进行了比较。为了简化讨论,将这三个特征分别表示为PSSM、HMM和word2vec。不同特征的结果如表3所示。在三个单独的特征中,PSSM的表现优于其他两个特征,表明进化信息对于预测肽的二级结构更有效。该模型融合了PSSM特征、HMM特征和多肽序列嵌入的多视点学习策略,与单个特征相比性能最好,说明不同的信息是相辅相成的,有效地提高了预测性能。为了更直观地理解特征,我们还进一步可视化了不同特征表示的特征空间分布,如图9所示。如图9所示,我们提出的多视图特征融合策略可以创建更好的特征空间,其中不同的结构状态被更清晰地分离,这进一步证明了多视图特征融合策略对提高特征表示能力是有效的。

表3.具有不同输入要素的模型的结果

图9.不同输入特征的主成分分析和t-SNE可视化:(A-D)分别表示PSSM、HMM、Word2vec和特征融合的PCA可视化结果;(E-H)分别表示PSSM、HMM、Word2vec和特征融合的t-SNE可视化结果。
个案研究
为了直观地比较我们的方法和现有方法的性能,我们随机选择了两个具有蛋白质数据库标识(PDB ID)的肽链-4jtm和1zt3,对这两个肽执行不同的二级结构预测方法。我们在图10中说明了预测结果,在图10中,我们分别给出了我们的方法PhD和Jpred的已知实验结构和预测结构。二级结构被映射到三级结构中,其中红色区域表示螺旋(H),黄色区域表示链(E),绿色区域表示线圈(C)。结果表明,与其他方法相比,用该方法预测的结构与实验结果更接近。特别是,我们的方法在局部连续序列区域上的性能比其他方法更好,进一步证实了我们的模型能够捕捉到更具区分性的局部区域信息。为此,我们可以得出结论,我们的方法比现有的方法更好。

图10。二级结构的可视化映射到三级结构为我们的方法和现有的方法,包括 phd 和 jpred。
网站
开发了一个用户友好的网络服务器,使读者能够更好地预测肽二级结构使用我们的最佳模型(e-state weighted 1.25)。该服务器使用 html、 javascript 和 java 作为前端开发,并安装在 ubuntu 企业 linux 服务器环境中。服务器采用最优序列作为输入,以文本格式显示二级结构。此外,我们的服务器可以进行一次多序列处理,如图11所示。此外,我们的代码和数据集可以在 mvirt https://github.com/massyzs/pssp- 免费下载。到目前为止,可以通过 http://server 访问 pssp-mvirt 服务器。Malab.cn/pssp- mvirt.
结论
在本研究中,我们发展了一种端到端的基于深度学习的肽二级结构预测方法—— pssp-mvirt。基准测试比较表明,我们的预测模型明显优于现有的方法,特别是在 acch 和 sov 上。此外,我们还研究了我们的模型在肽二级结构预测中的长度偏好性,并证明我们的模型在预测短肽时表现出更好的性能。此外,我们发现我们提出的多视点特征融合学习策略可以提高特征表示能力,从而提高预测性能。Pssp-mvirt 服务器可以为研究团体提供一种提高该方法性能的潜在途径。