利用TCGA癌症基因进行差异分析
TCGA癌症基因差异分析步骤
文章目录
- TCGA癌症基因差异分析步骤
-
- 1. 数据库下载
- 2. 将分散的文件转化为矩阵
- 3. 将矩阵id转化为基因名
- 4. 进行差异表达分析
1. 数据库下载
进入TCGA数据库官网,根据自己的需求下载各种癌症的数据库,全部勾选好对应的需求之后,下载解释文件(manifest),基因表达量文件(cart),临床数据(clinical),生物多样性数据(biospecimen),样品对应文件(metadata,tcga的id和病人id对应的文件)
最终下载的文件如上图所示。
2. 将分散的文件转化为矩阵
因为下载的文件大多都是json格式的文件,因此需要下载相应的perl包,这里用到的是json包。在perl package manager里面搜索json,然后下载json包即可,如图所示。
安装好之后就可以开始矩阵转化了。具体步骤如下所示:
- 首先解压gdc_download_20210518_081929.348052.tar文件,这里面都是基因在样本中的表达量数据。解压后的结果如图:
-
因为解压后的文件夹里面又包含了子文件,为了方便矩阵转化,需要将解压后的每个文件夹里面的文件放到同一个目录当中,这里我们采用perl语言,运行脚本来实现。脚本代码如下:
use strict; use warnings; my $newDir="files"; unless(-d $newDir) { mkdir $newDir or die $!; } my @allFiles=glob("*"); foreach my $subDir(@allFiles) { if(-d $subDir) { opendir(SUB,".\\$subDir") or die $!; while(my $file=readdir(SUB)) { if($file=~/\.gz$/) { `copy .\\$subDir\\$file .\\$newDir`; } } close(SUB); } }写好脚本之后,将脚本文件(.pl)和上面解压好的文件放在同一个目录下,然后打开powershell,并在powershell中进入到脚本所在的目录,然后输入perl putFilesToOneDir.pl即可运行脚本,最终会产生一个新的名为files文件夹,里面就是所有的解压后的文件了。如图所示

- 选中files文件夹里面的所有文件,然后右键,解压到当前文件夹,解压后的结果如图:

-
最后,将下载的metadata文件复制到files文件夹中,并新建名为mRNA_merge.pl脚本,脚本代码如下:
#!/usr/bin/perl -w use strict; use warnings; my $file=$ARGV[0]; #use Data::Dumper; use JSON; my $json = new JSON; my $js; my %hash=(); my @normalSamples=(); my @tumorSamples=(); open JFILE, "$file"; while() { $js .= "$_"; } my $obj = $json->decode($js); for my $i(@{$obj}) { my $file_name=$i->{'file_name'}; my $file_id=$i->{'file_id'}; my $entity_submitter_id=$i->{'associated_entities'}->[0]->{'entity_submitter_id'}; $file_name=~s/\.gz//g; if(-f $file_name) { my @idArr=split(/\-/,$entity_submitter_id); if($idArr[3]=~/^0/) { push(@tumorSamples,$entity_submitter_id); } else { push(@normalSamples,$entity_submitter_id); } open(RF,"$file_name") or die $!; while(my $line= ) { next if($line=~/^\n/); next if($line=~/^\_/); chomp($line); my @arr=split(/\t/,$line); ${$hash{$arr[0]}}{$entity_submitter_id}=$arr[1]; } close(RF); } } #print Dumper $obj open(WF,">mRNAmatrix.txt") or die $!; my $normalCount=$#normalSamples+1; my $tumorCount=$#tumorSamples+1; print "normal count: $normalCount\n"; print "tumor count: $tumorCount\n"; print WF "id\t" . join("\t",@normalSamples); print WF "\t" . join("\t",@tumorSamples) . "\n"; foreach my $key(keys %hash) { print WF $key; foreach my $normal(@normalSamples) { print WF "\t" . ${$hash{$key}}{$normal}; } foreach my $tumor(@tumorSamples) { print WF "\t" . ${$hash{$key}}{$tumor}; } print WF "\n"; } close(WF); 然后同样在powershell中,输入perl mRNA_merge.pl .\metadata.cart.2021-05-18.json,最终可以得到mRNAmatrix.txt这个文件,其表示的是tcga和ensembl id之间的对应关系,行名就是id,列名为矩阵

做到这里已经实现了矩阵转化,但在运行mRNA_merge.pl会输出如下结果:

表示正常的样本30个,癌症样本342个,对应在mRNAmatrix.txt中表示的就是第2-31列表示正常,32到最后表示癌症
3. 将矩阵id转化为基因名
虽然得到矩阵之后分析起来很方便,但是矩阵的id是ensembl的id,不易判断id对应的基因,因此这里需要将id转化为基因名字。具体步骤如下:
-
进入ensembl官网,下载ensembl数据,下载人类所对应的gtf文件,一般选择Homo_sapiens.GRCh38.104.chr.gtf文件下载。
-
将下载的gtf文件和矩阵文件mRNAmatrix.txt放在同一目录下
-
新建ensemblToSymbol.pl脚本,脚本代码如下:
use strict; use warnings; my $gtfFile=$ARGV[0]; my $expFile=$ARGV[1]; my $outFile=$ARGV[2]; my %hash=(); open(RF,"$gtfFile") or die $!; while(my $line=) { chomp($line); if($line=~/gene_id \"(.+?)\"\;.+gene_name "(.+?)"\;.+gene_biotype \"(.+?)\"\;/) { $hash{$1}=$2; } } close(RF); open(RF,"$expFile") or die $!; open(WF,">$outFile") or die $!; while(my $line= ) { if($.==1) { print WF $line; next; } chomp($line); my @arr=split(/\t/,$line); $arr[0]=~s/(.+)\..+/$1/g; if(exists $hash{$arr[0]}) { $arr[0]=$hash{$arr[0]}; print WF join("\t",@arr) . "\n"; } } close(WF); close(RF); -
打开powershell,输入perl ensemblToSymbol.pl Homo_sapiens.GRCh38.104.chr.gtf mRNAmatrix.txt mRNA.symbol.txt
(PS: mRNA.symbol.txt为输出文件,名字可以自己随便起)
-
最终得到的结果如图:
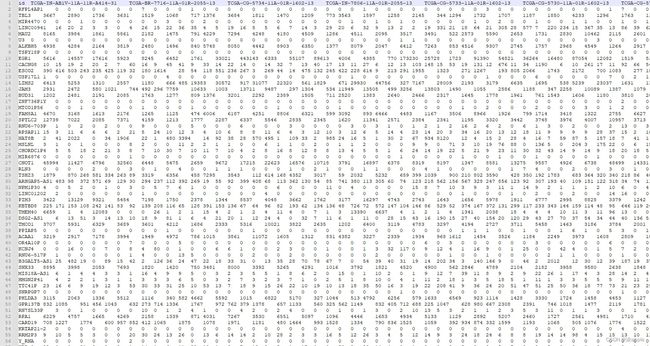
可以看到此时第一列就转化为基因名字了。
4. 进行差异表达分析
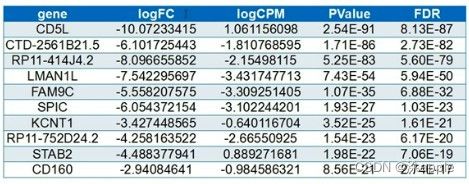
进行差异分析就是要得到这张表,第二列表示基因的fold change值,表示正常组织和癌组织的差异倍数的log值;第三列每一百万的抗体数,也是取了log值;第四列为p值,如果p=0表示基因在正常组织和癌症组织之间没差异,p越小,表示基因在正常组织和癌症组织有差异的概率越大;第五列表示错误发现率即False Discovery Rate,它是对p值的一个校正,也就是校正后的p值。
一般在差异表达分析时,选择logFC>1或logFC<-1(表示差异倍数在两倍以上),FDR<0.05的基因,这样的基因具有显著的差异水平
这里采用R语言的edger包做差异表达分析,代码如下:
source("http://bioconductor.org/biocLite.R") source("https://bioconductor.org/biocLite.R")
biocLite("edgeR")
install.packages("gplots")
foldChange=1 #如果筛选出来的样品过多,可以调大foldchange值或者调小padj即p值
padj=0.05
setwd("E:\\tcga3\\tcga_diff") #设置工作目录即mRNA.symbol.txt所在的路径
library("edgeR")
rt=read.table("mRNA.symbol.txt",sep="\t",header=T,check.names=F) #改成自己的文件名
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)
data=data[rowMeans(data)>1,]
#group=c("normal","tumor","tumor","normal","tumor")
group=c(rep("normal",30),rep("tumor",342)) #按照癌症和正常样品数目修改
design <- model.matrix(~group)
y <- DGEList(counts=data,group=group)
y <- calcNormFactors(y)
y <- estimateCommonDisp(y)
y <- estimateTagwiseDisp(y)
et <- exactTest(y,pair = c("normal","tumor"))
topTags(et)
ordered_tags <- topTags(et, n=100000)
allDiff=ordered_tags$table
allDiff=allDiff[is.na(allDiff$FDR)==FALSE,]
diff=allDiff
newData=y$pseudo.counts
write.table(diff,file="edgerOut.xls",sep="\t",quote=F)
diffSig = diff[(diff$FDR < padj & (diff$logFC>foldChange | diff$logFC<(-foldChange))),]
write.table(diffSig, file="diffSig.xls",sep="\t",quote=F)
diffUp = diff[(diff$FDR < padj & (diff$logFC>foldChange)),]
write.table(diffUp, file="up.xls",sep="\t",quote=F)
diffDown = diff[(diff$FDR < padj & (diff$logFC<(-foldChange))),]
write.table(diffDown, file="down.xls",sep="\t",quote=F)
normalizeExp=rbind(id=colnames(newData),newData)
write.table(normalizeExp,file="normalizeExp.txt",sep="\t",quote=F,col.names=F) #输出所有基因校正后的表达值(normalizeExp.txt)
diffExp=rbind(id=colnames(newData),newData[rownames(diffSig),])
write.table(diffExp,file="diffmRNAExp.txt",sep="\t",quote=F,col.names=F) #输出差异基因校正后的表达值(diffmRNAExp.txt)
heatmapData <- newData[rownames(diffSig),]
#volcano
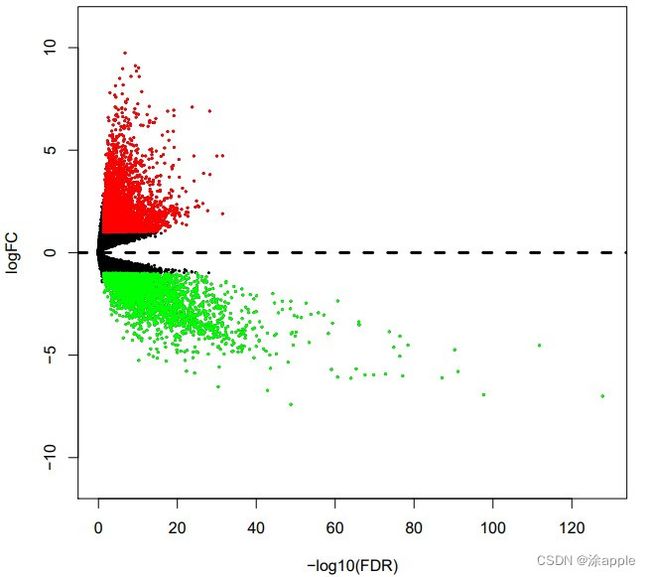
pdf(file="vol.pdf")
xMax=max(-log10(allDiff$FDR))+1
yMax=12
plot(-log10(allDiff$FDR), allDiff$logFC, xlab="-log10(FDR)",ylab="logFC",
main="Volcano", xlim=c(0,xMax),ylim=c(-yMax,yMax),yaxs="i",pch=20, cex=0.4)
diffSub=allDiff[allDiff$FDRfoldChange,]
points(-log10(diffSub$FDR), diffSub$logFC, pch=20, col="red",cex=0.4)
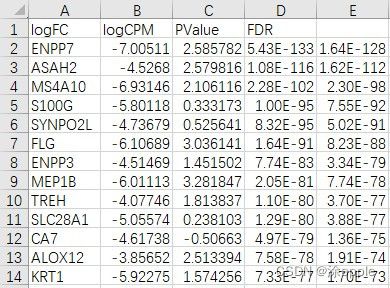
diffSub=allDiff[allDiff$FDR 最终即可得到差异表格,热图和火山图等,如图所示:

下图为基因差异表格:
下图为热图:

下图为火山图: