【基因芯片】差异表达分析的基本原理与方法
【基因芯片】差异表达分析的基本原理与方法
原文链接 https://mp.weixin.qq.com/s?__biz=Mzg4MDc2MzUwMg==&mid=2247483703&idx=1&sn=61c9d3ec29d027fe17a5098b30611342&chksm=cf717636f806ff205425bd30fba444758c8b2a3e56bdcf733b9706007612420ea9a35b39e1c0#rd
微信公众号,生信Cat
引言
生物芯片产生于1991年,其前身是分子点阵杂交技术。芯片数据在前十年使用较为广泛,但自从RNA-seq技术出现后便迅速没落,鲜有使用。不过目前GEO数据库中仍有大量的芯片数据可供挖掘,因此今天来讨论一下芯片数据的数据预处理和差异分析的基本原理。
01
—
基因表达的测定原理----杂交测序
杂交测序是指:提取样本总mRNA后,通过反转录过程获得标记荧光的核酸序列,然后与探针进行杂交反应后,再将未互补结合的片段洗去。对基片进行激光共聚焦扫描,测定芯片上各点的荧光强度来推算样品中各种基因的表达量。
最常见的两种技术分别为cDNA芯片和Affymetrix公司的寡核苷酸芯片。前者探针是cDNA,后者则是寡核苷酸。
02
—
数据预处理(pre-procession)
基因芯片的数据预处理主要包括数据提取,数据过滤,补缺失值,对数转化,标准化处理、
1,数据提取
主要目的是将高通量的荧光信号转化成基因表达数据,形成原始表达矩阵,包括探针ID,样本中每个基因对应的表达水平(荧光强度)。该矩阵可以用于后续分析。
2,数据过滤
基因芯片数据有很高的背景噪音及假阳性,数据过滤便是用于应对背景噪音的处理步骤,一般是去除表达量很小、负值或明显的噪音数据。通常是删除或赋予统一数值。
3,补缺失值
缺失值主要有两种类型,一种是随机缺失,即表达矩阵中的数据缺失与基因表达值的高低无关,是由其他因素造成的,如污染,杂交失败等,数据补缺适合这类情况。另一种是非随机缺失,数据的缺失与表达丰度有关,基因表达丰度过低或过高都有可能出现数据缺失,对于这种缺失没有较好的处理方法。
目前的缺失处理主要有三种方法
-
简单补缺法,用0,1,每行或每列的均值作为缺失的可能信号值
-
K近邻法,对于含有缺失值的基因i 的k个邻居,设X1j,X2j,······ ,Xkj 分别为基因 i 的k个邻居基因在第 j 个样本中的表达值。常用于定义邻居基因的距离函数有欧氏距离或相关系数。用邻居基因在该样本中的加权平均评估缺失值。Wg为权重系数,由邻居基因g与基因 i 的距离决定。
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-Jq2LNhFH-1650849183581)(data:image/gif;base64,iVBORw0KGgoAAAANSUhEUgAAAAEAAAABCAYAAAAfFcSJAAAADUlEQVQImWNgYGBgAAAABQABh6FO1AAAAABJRU5ErkJggg==)]
-
回归法:与K近邻法类似,回归法用回归模型预测缺失值,然后再加权平均。
4,对数转化
一般认为基因芯片的原始数据呈偏态分布,通过对数转化后可以使数据近似服从正态分布,从而简化后续分析。通常取以2为底的对数。
5,标准化 归一化 (normalization)
因为在细胞中有一系列稳定表达的基因,管家基因和人工合成的控制基因,可以认为这些基因的荧光强度值的差异主要是由系统误差造成的,所以常运用这些稳定表达的基因作为参照基因,修正其他基因荧光强度的系统误差。对于不同的芯片平台,制作原理不同,引入的系统误差不同,标准化的方式略有差异。
标准化与归一化的区别回头专门写个帖子讲QAQ,查了很多资料发现这俩说啥的都有,这里先不要纠结啦~
03
—
差异分析
差异分析是为了识别差异基因,在排除实验干扰,误差等因素后,得到具有统计学意义同时具有生物学意义的基因集合。换句话说,就是判断组间(处理)差异是否显著大于组内(误差)差异 。
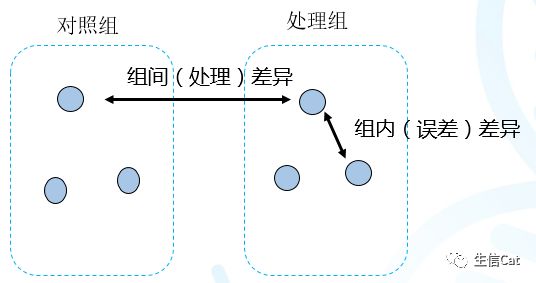
目前常用的差异分析方法有倍数分析,假设检验,建模分析。
一,倍数分析(Fold Change 算法)
最传统的差异表达基因的鉴别方法是倍数法,该方法计算同一基因在两个条件下的表达水平的比值,如果变化比值超过一个常数,典型的阈值是2,由经验给出,则认为该基因的表达差异是显著的。如果有多次实验重复,则分别计算每次实验中两个条件下的基因表达的对应比值,再取均值。人们常常将其做 log2 转换,得到[-1,1]作为阈值,识别差异基因。
该方法简单,但没有考虑差异表达的统计显著性并且过于依赖分析人员的经验数值,因此局限较大。由于表达量低的基因较表达量高的基因更容易在两类间产生大的倍数变化,导致FC方法偏向于识别基础表达量低的基因作为差异基因。此外,表达量低的基因更容易因为检测误差的影响而产生大的波动从而被FC方法判断为差异基因
二,假设检验
假设检验的定义请自行查阅生物统计学书,此处不过多赘述。推荐《统计学(第三版)》张德存主编,2020年,科学出版社出版 。
1,参数方法
如果进行假设检验时总体的分布形式已知,需要对总体的未知参数进行假设检验,则称参数假设检验;若不清楚总体分布形式,需要对未知分布函数的形式及其特征进行假设检验则称非参数假设检验。在使用参数检验方法的时候,我们通常需要考虑数据的两个特征,即数据的总体分布和方差。
一般的,我们认为芯片数据符合正态分布(连续型),而RNA-seq数据符合泊松分布(离散型)。
-
-
t检验 StatQuest - 如何选择t检验
t检验主要用于样本含量较小(n<30),总体标准差未知的正态分布样本。它基于t分布理论来推断差异发生的概率,用于考察单个样本均值与总体均值之间的差异或两个均值之间的差异是否显著。
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-o1kpuSMf-1650849183583)(data:image/gif;base64,iVBORw0KGgoAAAANSUhEUgAAAAEAAAABCAYAAAAfFcSJAAAADUlEQVQImWNgYGBgAAAABQABh6FO1AAAAABJRU5ErkJggg==)]
-
-
对于两组数据而言,可以使用配对样本t检验,此处我们假设样本呈正态分布****。
-
其假设为
-

-
其计算公式为
-
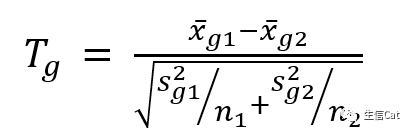
-
其中
-
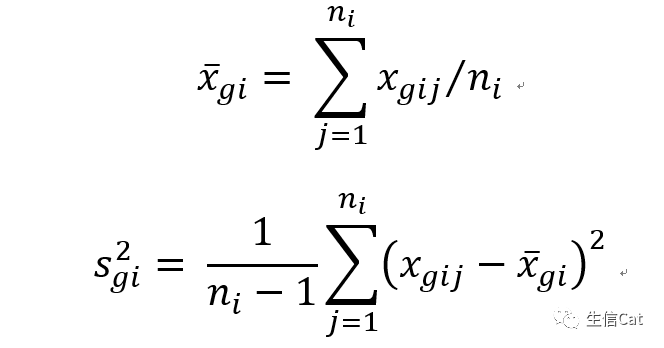
-

-
对于芯片数据,n值常常等于2,3,由于样本量小,总体方差被严重低估,使得T值较大,从而产生较高的假发现率(False Discorverey Rate ,FDR)。在T检验中,常使用0.01为显著水平。通过对T检验进行改进,得到更好的分析结果。
-

-
由于t检验要求数据呈现正态分布,所以公式中基因的表达值为测量值经过标准化后的值,反应的是两类样本间基因表达的倍数变化,也存在FC方法同样的偏向性。
-
此外,对基础表达量低的基因来说,一个微小变异程度(标准误)可能导致一个大的绝对 t 统计值,从而被识别为差异基因,即使在两类条件下这个基因的平均表达水平的差异很小。低表达的基因比高表达的基因更容易产生大的t统计量。
-
有研究指出,数据的信噪比会随着基因表达量的增高而降低,这就意味着,低表达的基因更容易受到噪声的影响而产生误差。因此,t检验同样倾向于识别表达水平低的基因作为差异基因。
-
-
方差分析
对于两组以上的数据比较,我们常采用方差分析。它将基因在样本之间的总变异分解为组间变异和组内变异两部分,通过方差分析的假设检验判断组间变异是否存在,如果存在则表明基因在不同条件下的表达有差异。分别计算总变异,组间变异,组内变异。
将变异除以自由度计算均方,消除自由度的影响
依据统计量F值,得到P值,判断基因表达是否有差异。
-
2,非参数方法(非参数检验)
-
SAM算法
SAM(Significance analysis of microarrays)算法用于微阵列基因表达谱数据识别差异基因 。SAM算法与t检验相似,但为了使具有较小标准误的基因不会被误判为差异基因,SAM在t统计量的分母中增加了校正值,提高了t检验的稳定性。计算公式如下,其中S0 为样本残差标准误的校正值:
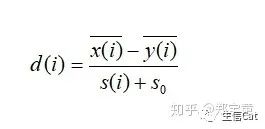
虽然SAM算法通过permutation算法计算出错误发现率(False Discovery Rate, FDR)来控制多重检验的错误率,降低了结果的假阳性率。但由于SAM算法是以t检验为基础,但它依旧存在与t检验相似的问题:偏向于识别在两类样本中表达水平低但倍数变化大的基因为差异基因。
三,建模分析
通过确定两个条件下的模型参数是否相同来判断表达差异的显著性,例如贝叶斯方法。(略)
参考资料 :
[1] 李霞主编,《生物信息学》(卫生部八年制规划教材),2015年,人民卫生出版社
[2] https://zhuanlan.zhihu.com/p/388984969
[3] 李春喜等,《生物统计学》第五版,2013,北京:科学出版社
[4] 刘伟等,《生物信息学》第二版,2018,电子工业出版社
[5] 吕晓玲 黄丹阳 著 《数据科学统计基础》第一版,北京:中国人民大学出版社出版,2021.1[6] https://zhuanlan.zhihu.com/p/50526813【基因芯片】差异表达分析的基本原理与方法