RNA 1. SCI 文章中读取 GEO 数据
依稀记得10年前一个样本的RNA-SEQ费用还是蛮高的,而现在就是洒洒水啦,所以这种转录组的数据也已经成为文章的主流,基本上就是WES结合RNA,但是个人感觉,真的能关联到突变基因和表达的水平上去的,还是很困难的,除非您运气好到爆表,但是表观遗传如甲基化,乙酰化等,一致与表达紧密相关,毕竟他们都算是不改变DNA编码而只是在其链上做些装饰,就像好多美女都会化各种妆容,但是一旦卸了妆,还是那个素颜的自己,。
这次我们就说说转录组那些事,基本原来到单细胞转录组,我们该怎么做,以及涉及的软件。
01 NGS的****RNA测序的原理
RNA 不像 DNA 那样可以测到完整的基因组,文库制备方法RNA-seq通常从 total RNA samle 制备开始,然后是核糖体去除步骤。在标准的NGS建库之前,total RNA sample会被转换为 cDNA。RNA-Seq 聚焦于 mRNA, smallRNA, noncodingRNA, microRNAs,可以通过在cDNA 合成前附加的分离或富集步骤来实现。如下图所示:
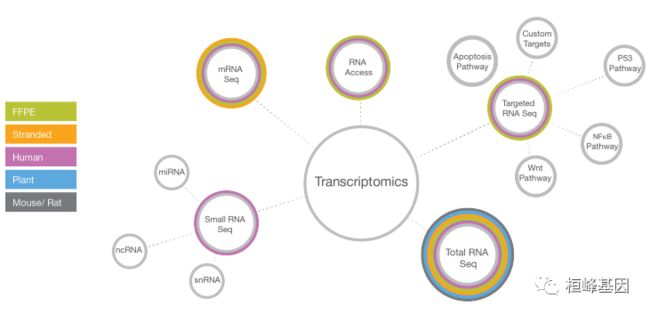
RNA 的测序属于常规测序,遵循测序的常规操作即可,一般都是使用 illumina 测序仪,如下:

当然这是NGS的测序,但是还有其他平台的测序,比如GEO 作为各种高通量实验数据的公共存储库,主要包括这几类平台的数据:
-
基于单通道和双通道微阵列的实验;
-
质谱蛋白质组学数据;
-
高通量测序数据。
02 基因表达量的计算方法
目前计算表达量已经有四种方式来计算,那么哪种方式最适合,那只能说咱们也就跟下潮流,之前大家认可的 RPKM 也让各路专家们认定为不准确,但是甭管怎么说,存在就是还有到了,还需要仔细推敲其中的原理。四种计算方式如下:
- RPKM (Reads Per Kilobase of exon model per Million mapped reads)
RPKM(推荐软件,Range) 的计算公式:
RPKM= total exon reads/ (mapped reads (Millions) * exon length(KB));
total exon reads:某个样本mapping到特定基因的外显子上的所有的reads;
mapped reads ( Millions ) :某个样本的所有reads总和;
exon length( KB ):某个基因的长度(外显子的长度的总和,以KB为单位)。
比如一个样本中一个基因的RPKM等于落在这个基因上的总的read数(total exon reads)与这个样本的总read数(mapped reads (Millions))和基因长度(exon length( KB )) 的乘积的比值。
- FPKM (Fragments Per Kilobase of exon model per Million mapped fragments)
-
FPKM:每千个碱基的转录每百万映射读取的fragments,主要是针对pair-end测序表达量进行计算。
-
FPKM和RPKM的区别就是一个是fragment,一个是read。
- RPM/CPM (Reads/Counts of exon model per Million mapped reads)
-
RPM/CPM:每百万映射读取的reads;
-
RPM=total exon reads / mapped reads (Millions);
-
total exon reads:某个样本mapping到特定基因的外显子上的所有的reads;
-
mapped reads (Millions) :某个样本的所有reads总和。
- TPM (Transcripts Per Kilobase of exonmodel per Million mapped reads)
-
TPM:每千个碱基的转录每百万映射读取的Transcripts),优化的RPKM计算方法,可以用于同一物种不同组织的比较;
-
TPMi={( Ni/Li )*1000000 } / sum( Ni/Li+………+ Nm/Lm );
-
Ni:mapping到基因i上的read数;
-
Li:基因i的外显子长度的总和。
在一个样本中一个基因的TPM:先对每个基因的read数用基因的长度进行校正,之后再用校正后的这个基因read数(Ni/Li)与校正后的这个样本的所有read数(sum(Ni/Li+………+ Nm/Lm))求商。由此可知,TPM概括了基因的长度、表达量和基因数目。
03 GEO 数据特征
GEO (Gene Expression Omnibus),是由美国国立生物技术信息中心 NCBI 创建并维护的基因表达数据库,创建于2000年,收录了世界各国研究机构提交的高通量基因表达数据,也就是说只要是目前已经发表的论文,论文中涉及到的基因表达检测的数据都可以通过这个数据库中找到。
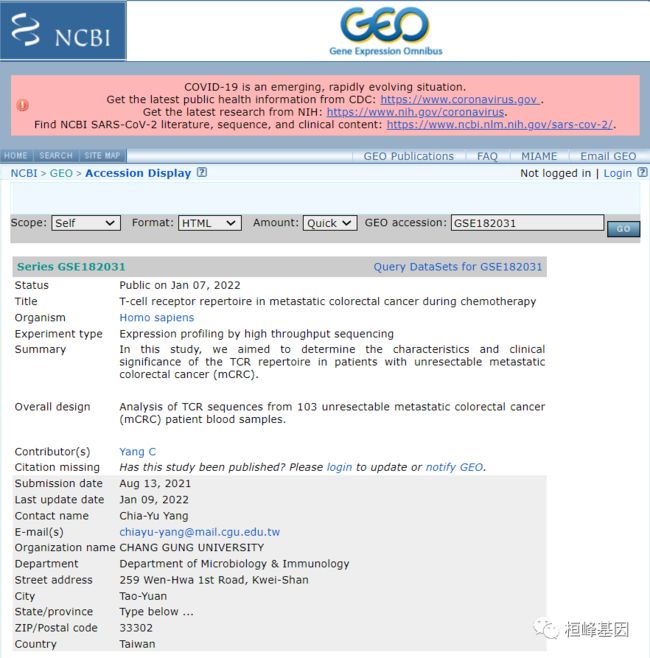

这个数据库是免费的!
在 GEO 最基本的组织层面,有四种基本实体类型。前三个(样本,平台和系列)由用户提供; 第四,数据集由 GEO 工作人员根据用户提交的数据进行编译和策划。
SCI 文章可以有一个或多个GSE数据集,一个GSE里可以有一个或多个GSM样本。多个研究的GSM样本可以根据研究目的整合为一个GDS,每个数据集有着自己对应的芯片平台(GPL),一个GSE里可能有多个平台测出的数据。因此针对不同平台的数据结果需要我们使用正确的代码进行读取,首先看第一类微阵列的数据,主要有几大平台数据。
我们以结直肠癌为例子,选择样本量最多的那个数据集,如下:
-
数据集:GSE41258
-
测序平台: Affymetrix Human Genome U133A Array
-
样本量:390
-
网站:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41258

04 GEO 数据获取基因表达矩阵
现有的基因芯片种类很多,但是重要而且常用的芯片并不多!一般分析芯片数据都需要把探针的ID切换成基因的ID,最终目的就是为了获得 symbol,一般有三种方法可以得到芯片探针与gene的对应关系。
-
基因芯片的厂商的官网直接去下载;
-
一种是直接用bioconductor的包;
-
一种是从NCBI里面下载文件来解析!
用R的bioconductor包来批量得到芯片探针与gene的对应关系!一般重要的芯片在R的bioconductor里面都是有包的,用一个R包可以批量获取有注释信息的芯片平台,这里我只选取人的数据,如下:
number gpl organism bioc_package
1 GPL80 Homo sapiens hu6800
2 GPL91 Homo sapiens hgu95av2
3 GPL92 Homo sapiens hgu95b
4 GPL93 Homo sapiens hgu95c
5 GPL94 Homo sapiens hgu95d
6 GPL95 Homo sapiens hgu95e
7 GPL96 Homo sapiens hgu133a
8 GPL97 Homo sapiens hgu133b
9 GPL98 Homo sapiens hu35ksuba
10 GPL99 Homo sapiens hu35ksubb
11 GPL100 Homo sapiens hu35ksubc
12 GPL101 Homo sapiens hu35ksubd
13 GPL201 Homo sapiens hgfocus
14 GPL570 Homo sapiens hgu133plus2
15 GPL571 Homo sapiens hgu133a2
16 GPL886 Homo sapiens hgug4111a
17 GPL887 Homo sapiens hgug4110b
18 GPL1352 Homo sapiens u133x3p
19 GPL1708 Homo sapiens hgug4112a
20 GPL2891 Homo sapiens h20kcod
21 GPL3921 Homo sapiens hthgu133a
22 GPL4191 Homo sapiens h10kcod
23 GPL5689 Homo sapiens hgug4100a
24 GPL6097 Homo sapiens illuminaHumanv1
25 GPL6102 Homo sapiens illuminaHumanv2
26 GPL6244 Homo sapiens hugene10sttranscriptcluster
27 GPL6947 Homo sapiens illuminaHumanv3
28 GPL8300 Homo sapiens hgu95av2
29 GPL8490 Homo sapiens IlluminaHumanMethylation27k
30 GPL10558 Homo sapiens illuminaHumanv4
31 GPL11532 Homo sapiens hugene11sttranscriptcluster
32 GPL13497 Homo sapiens HsAgilentDesign026652
33 GPL13534 Homo sapiens IlluminaHumanMethylation450k
34 GPL13667 Homo sapiens hgu219
35 GPL15380 Homo sapiens GGHumanMethCancerPanelv1
36 GPL15396 Homo sapiens hthgu133b
37 GPL17897 Homo sapiens hthgu133a
GEOquery 是专门用于读取 GEO 数据库的数据,用起来很方便,我们这里就是利用其有缺的特点成功获得了 prob 和样本的表达矩阵,但是这并不是目的,还需要我们将prob转换为基因symbol,这才是我们想要的结果,如下:
rm(list = ls())
## 魔幻操作,一键清空~当前环境中对象全部删除
options(stringsAsFactors = F)
#在调用as.data.frame的时,将stringsAsFactors设置为FALSE可以避免character类型自动转化为factor类型
f='GSE41258_eSet.Rdata'
#把GSE41258_eSet.Rdata赋值给f,方便后面流程化处理
##根据数据集不同修改相应的GSE号
#BiocManager::install("GEOquery")
library(GEOquery)
# 这个包需要注意两个配置,一般来说自动化的配置是足够的。
#Setting options('download.file.method.GEOquery'='auto')
#Setting options('GEOquery.inmemory.gpl'=FALSE)
if(!file.exists(f)){
gset <- getGEO('GSE41258', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
save(gset,file=f) ## 保存到本地
}
##这是一个函数,利用包将数据集的表达信息下载下来,赋值给了gset,而不下载注释信息和平台信息,并保存到本地,文件名为f。
load('GSE41258_eSet.Rdata')
## 载入数据
class(gset)
#查看数据类型
length(gset)
##看一下有几个元素
gset[[1]]
#取第一个元素
class(gset[[1]])
#查看该元素的数据类型
# 因为这个GEO数据集只有一个GPL平台,所以下载到的是一个含有一个元素的list
a=gset[[1]]
##取出第一个元素赋值给一个对象a
dat=exprs(a)
#a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数,该函数得到表达矩阵
#现在 得到的dat就是一个表达矩阵,只不过基因的ID是探针名
dim(dat)
#看一下dat这个矩阵的维度
dat[1:5,1:5]
#查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
#这个表达矩阵是已经log之后的,表达量一般是0-10左右,如果是原始芯片表达的信号值一般是几千到一万,则需要log处理。
pd=pData(a)
#通过查看说明书知道取对象a里的临床信息用pData
View(pd)
## 查看一下,挑选一些感兴趣的临床表型,这里我们欲得到其分组title信息。
library(stringr)
#运行一个字符分割包
group_list=str_split(pd$title,' ',simplify = T)[,4]
#抽取title一列,按照空格分割,取第四个元素即Control和Vemurafenib
table(group_list)
#看一下两个分组各有几个
我们根据数据集对应的 R 包为 hgu133a.db,下载并安装,获取基因探针对应的基因 symbol,最后我们获得了一个行为样本,列为基因的基因表达的矩阵,用于后续的分析,如下:
########
#BiocManager::install("hgu133a.db")
library(hgu133a.db)
##运行这个包
ls("package:hgu133a.db")
#对这个包进行探索,看一下有多少元素
##找到有SYMBOL的那个元素就是我们需要的对应关系
##例如:[34] "hgu133aSYMBOL"
ids=toTable(hgu133aSYMBOL)
#toTable这个函数:通过看hgu133plus2.db这个包的说明书知道提取probe_id(探针名)和symbol(基因名)的对应关系的表达矩阵的函数为toTable。并赋值给ids
##这时候我们得到19825个探针对应的基因名。
###刚才我们的表达矩阵中是33297个基因探针,这就意味着刚才的表达矩阵中可能存在多个探针重复对应一个基因名。这就需要我们对数据进行进一步筛选、处理。
#为查看前六行
colnames(ids)=c('probe_id','symbol')
#将列名统一改为'probe_id','symbol'方便后续统一操作。
length(unique(ids$symbol))
tail(sort(table(ids$symbol)))
table(sort(table(ids$symbol)))
ids=ids[ids$symbol != '',]
ids=ids[ids$probe_id %in% rownames(dat),]
##%in%用于判断是否匹配,然后取匹配的几行,去掉无法匹配的信息。
dat[1:5,1:5]
dat=dat[ids$probe_id,]
#取表达矩阵中可以与探针名匹配的那些,去掉无法匹配的表达数据,这时只剩下19825个探针及表达信息,其余已被剔除。
ids$median=apply(dat,1,median)
#ids新建median这一列,列名为median,同时对dat这个矩阵按行操作,取每一行的中位数,将结果给到median这一列的每一行
ids=ids[order(ids$symbol,ids$median,decreasing = T),]
#对ids$symbol按照ids$median中位数从大到小排列的顺序排序
##即先按symbol排序,相同的symbol再按照中位数从大到小排列,方便后续保留第一个值。
##将对应的行赋值为一个新的ids,这样order()就相当于sort()
ids=ids[!duplicated(ids$symbol),]
#将symbol这一列取取出重复项,'!'为否,即取出不重复的项,去除重复的gene ,保留每个基因最大表达量结果.最后得到18832个基因。
dat=dat[ids$probe_id,]
#新的ids取出probe_id这一列,将dat按照取出的这一列中的每一行组成一个新的dat
rownames(dat)=ids$symbol
#把ids的symbol这一列中的每一行给dat作为dat的行名
head(dat)
##至此我们就得到了该数据集的表达矩阵,最后将结果保存。
write.csv(dat,file="GSE41258.expressionmetrix.csv")

到处我们已经获得了想要的数据集,整理好数据,等待下次我们将进行差异基因表达的分析。敬请期待
关注微信公众号:桓峰基因
扫码进群免费答疑,桓峰助力 SCI 高分,找桓峰,是您最好的选择!

Reference:
-
Sheffer M, Bacolod MD, Zuk O, Giardina SF et al. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci U S A 2009 Apr 28;106(17):7131-6.
-
Martin ML, Zeng Z, Adileh M, Jacobo A et al. Logarithmic expansion of LGR5+ cells in human colorectal cancer. Cell Signal 2018 Jan;42:97-105.
桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
33篇原创内容
公众号
