单细胞分析概览
目录
- 前置内容
-
- 前提概念:细胞,基因,蛋白质的关系
- Alpha fold中的PAE
- 单细胞数据上游来源
- 质量控制
- 去除细胞效应和基因效应
- 降维与聚类分群
- 细胞亚群注释
- 细胞亚群继续分群
- 细胞亚群比例比较
- 细胞轨迹分析(需要具有背景知识)
单细胞分析是重要的,应用:癌症研究,发育生物学,免疫学
前置内容
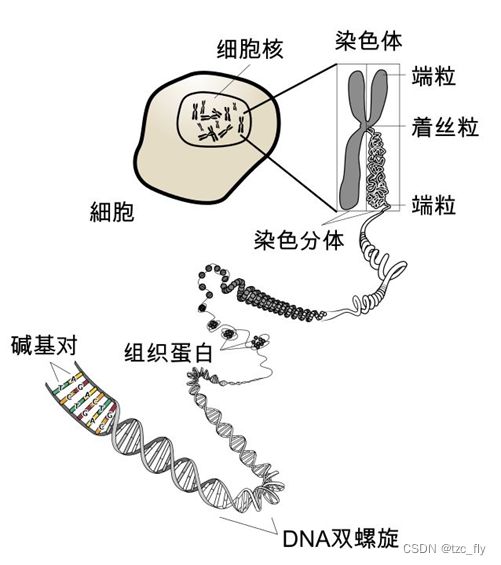
前提概念:细胞,基因,蛋白质的关系
蛋白质与多肽的区别在于它们的结构、功能和内涵不同。 多肽指的是,多个氨基酸脱水缩合形成的线性氨基酸链。 蛋白质,可能只具有一条多肽链,也可能是由多条多肽链构成。
蛋白质是由氨基酸(分子中具有氨基和羧基的一类有机化合物)构成的生物大分子;
基因:是遗传的基本单元,是携带遗传信息的DNA片段。可以简单理解为一段DNA链中比较特殊的某段。
染色体包含DNA,某些蛋白质;
DNA包含基因;
某些蛋白质它的功能是组成染色体,某些蛋白质它承担了生物生命活动的其它功能。
基因与蛋白质的关系:DNA中的基因指导蛋白质的合成:
Alpha fold中的PAE
当预测结构和真实结构在残基 y 上对齐时(将预测结构的残基和真实结构的残基对齐),位置 (x, y) 处的颜色表示 AlphaFold 在残基 x 处的期望位置误差:

举例:
深绿色是好的(低错误),浅绿色是坏的(高错误)。 例如,在残基 300 上对齐预测结构与真实结构,根据PAE plot我们可以知道预测结构:
- 我们对残基 200 的预测相对位置充满信心;
- 我们对残基 600 的预测相对位置没有信心;

单细胞数据上游来源
对科学机构来说:基因测得多更有价值,因为细胞测得多可以被商业公司取代。
上游数据来源:主要是10x数据和Smart-seq2数据。
- 每个10x的样本(来自一个病人的单细胞数据):该样本的表达矩阵由3个文件构成;
- Smart-seq2的每个样本的表达矩阵只需要一个文件描述。
通常一个样本就需要很多资金购买;10x的表达矩阵由3个文件构成:因为表达矩阵0值很多,稀疏,换成3个文件去描述可以节省空间。
质量控制
质量控制可以过滤不合格的细胞和基因。
细胞过滤(看文库大小和检测到的基因数量):给全部细胞检测到的基因数量绘制boxplot,看看哪些细胞检测到的基因数量偏多或者偏少,这些细胞需要去除。
基因过滤:1.通常,表达矩阵里的基因根据gtf文件注释,表达矩阵中覆盖了gtf所有基因,但通常大量基因在细胞里是没有的,所以表达量为0,需要去除。2.设置阈值,一个基因表达多少才算是表达,一个基因在多少个细胞里表达过才保留。
过滤线粒体核糖体基因(质量控制的选做步骤):这是一个很难把握的工作,需要结合自己项目的情况来做。不过通常有以下策略:1.粗暴去除所有线粒体核糖体基因,直接去除包含”MT-”开头的基因。2.选择阈值去除高表达量的细胞,阈值很大程度上取决于对自己项目的了解程度,因为不同器官组织提取的单细胞,线粒体基因平均水平不一样。
去除细胞效应和基因效应
对表达矩阵进行归一化和标准化(去除细胞效应和基因效应):在获取到干净的表达矩阵后,并且在进行下游分析之前,需要对表达矩阵标准化。
1.首先去除细胞效应:不同细胞的测序量不一样,对于同一个基因,在不同细胞里,虽然看到的表达量是一样的,但是背后细胞整体测序数据量的差异其实反而说明了这个基因在不同细胞表达量其实是有差异的。所以要在细胞维度进行标准化。
2.去除基因效应:在绘制热图的时候会需要使用,因为个别基因表达量过高,在热图里面会一枝独秀,实际上我们并不会关心不同基因的表达量高低,我们仅仅是想看指定基因在不同细胞中的高低而已,所以我们沿着基因维度标准化。
3.取log降低表达量的离散程度:原始的raw counts矩阵是一个离散型的变量,离散程度很高。有的基因表达丰度比较高,counts数为10000,有些低表达的基因counts可能10,甚至在有些样本中为0。其实大量的基因在很多细胞的表达量都是0,如果是10x数据,甚至会出现一个表达矩阵里面97%的数值都是0 的现象。如果对表达量取一下log10,发现10000变成了4,10变成了1,这样之前离散程度很大的数据就被集中了。有时当表达量为0时,取log会出现错误,可以log(counts+1)来取log值。当x=1时,所有的log系列函数值都为0。这样原本表达量为0的值,取log后仍为0。
降维与聚类分群
首先需要降维:1.硬过滤,根据一些统计指标(sd,mad,vst),筛选表达矩阵中的高变基因,高变异基因就是highly variable features(HVGs),就是在细胞与细胞间进行比较,选择表达量差别最大的;2.可以采用PCA线性降维,将1.处理后的表达矩阵降维到十几个维度(通常top20主成分)。
然后进行聚类:聚类算法各种各样。
聚类结果我们希望对其可视化(二维平面的散点图),这个可视化其实是使用降维算法完成的,通常是t-sne和umap。
t-sne相对于PCA有更高的区分度,不同细胞类型分得开;
t-sne保留局部结构,相类似的细胞聚集在一起,同时不会过于重叠扎堆;
相对于UMAP,t-sne不能很好的展示全局结构;
经典的tSNE算法不适用于较大的数据;
鉴于tSNE的缺点,近年来umap逐渐在单细胞数据的可视化方法中占据位置。umap相对tSNE来说,保留了数据的全局结构,而且大数据的运行时间较短。
细胞亚群注释
目前,细胞亚群注释依赖标记基因。降维与聚类并不是单细胞分析的重点,我们要的是得到基于聚类结果的不同细胞亚群命名,给出生物学的解释。比如文章” Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA”对PBMC(外周血单核细胞)分群得到13个细胞类群并细致决定了亚群的名字。

在” dropClust: efficient clustering of ultra-large scRNA-seq data”中就已经对PBMC的不同细胞亚群与标记基因(Markers)建立了关联:
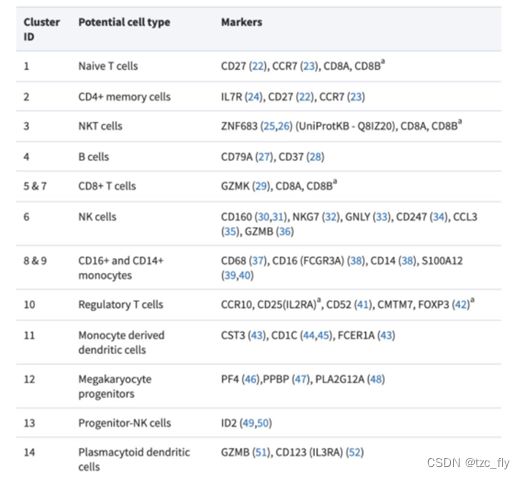
目前已知可靠的细胞中的标记基因可以自己查阅文献发现。
在数据分析过程中,我们可以对比不同的细胞分群,观察基因的热图,从而发现标记基因。把细胞亚群的基因表达值取一个统计量(平均值或者中位值)来作为该基因在该细胞亚群的表达量。所以热图展现多个标记基因在全部细胞亚群的表达量热图,也不会拥挤:
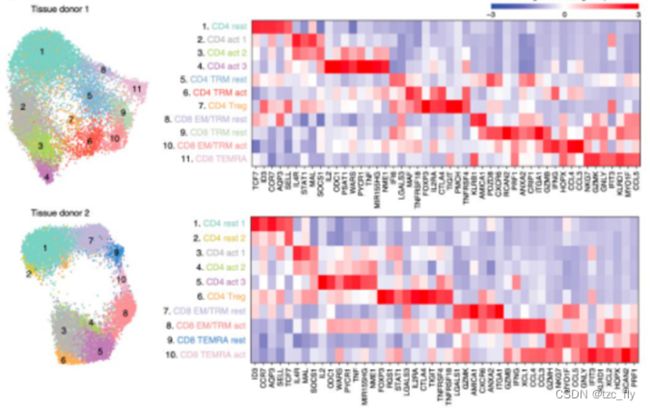
但是,如果要展现多个标记基因在全部细胞的表达量,而不仅仅是细胞亚群,热图就会很拥挤,如下所示:

细胞亚群继续分群
理论上细胞亚群是可以无限划分的,因为世界上没有两个一模一样的细胞,关键是要把握一个度,什么样的差异可以判定为不同细胞亚群,什么样的差异是可以容忍的细胞类群内部异质性。
有一个策略就是找出主要因素和次要因素。主要因素划分为主要亚群,比如外周血PBMC里面的T,B细胞当然是不同亚群,但是T细胞里面还可以继续划分:CD4或者CD8的T细胞,甚至继续划分, 如下图所示:

现在10x单细胞转录组技术一个样本出来3~8K细胞的数据,很多课题都是十几个以上样本,所以容易到十万多个单细胞数量级,它们走聚类分群对技术资源的消耗当然是很可观的,而且,拿到的细胞亚群数量也是惊人的。比如分成了近100个细胞亚群。
实际上,细胞二维散点图,是没办法写出全部细胞亚群的生物学功能定义,我们通常是把主要细胞亚群标记上去。然后每个细胞亚群再进行继续分群。只不过是在同一个散点图上面展示。
注意,如果子亚群划分过深入,导致命名的时候不一定都是有很明确生物学功能的。
细胞亚群比例比较
如果要比较细胞亚群比例,就必须要有多个样本。因为我们比较的是不同样本之间,相同细胞亚群的比例分布。
可以看到下面是2个单细胞转录组样本,它们的批次效应已经去除,tSNE图如下(红细胞和血小板):
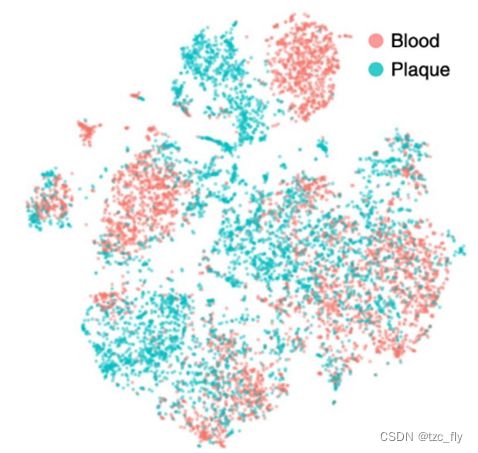
根据聚类分群结果,对各个样本的细胞亚群命名后,重新拆分开来看两个样本的不同细胞亚群的比例,如下:

理论上分群后,注释了亚群名称,就可以看到细胞亚群在这不同样本之间的比例差异。
细胞轨迹分析(需要具有背景知识)
常见的单细胞轨迹研究方法包括基于树图的拟时分析及RNA 速率分析(RNA velocity)等。
RNA 速率分析通过拟合前体(未剪接的)和成熟(剪接的)mRNA 丰度之间的比值来获得基因特异性速度,得出可能的细胞状态变化,从而追溯细胞的起源和潜在的命运,RNA速率分析不需要指定起点和终点。
对于不同的轨迹分析算法,基于树图的拟时序分析需要研究人员根据潜在细胞分化轨迹及关键基因的表达,确认细胞分化的起点和终点。
RNA速率分析直接提供了细胞轨迹分析的起止点,可以对未知分化关系的细胞进行分析。
研究人员可以直接选择具有潜在分化关系的亚群或者自己关注的细胞亚群进行树图拟时序分析。而对于不确定分化关系的细胞,研究人员可以通过RNA速率分析对所有细胞进行分析,筛选出存在分化关系的亚群,进一步进行树图拟时序分析,挖掘分化轨迹的关键亚群和基因。
应用:细胞轨迹分析可以验证已知的细胞分化关系,揭示细胞发育轨迹,挖掘肿瘤免疫细胞动态变化,挖掘分化关键的细胞亚群及基因。