5. 生信技能树——GEO转录组RNA_seq_GSE162550
和生信技能树GEO转录组“GSE150392“分析类似,唯一区别就是在数据处理和ID转换这一环节略微有区别
1.数据下载
最方便的是xena。可以网页下载,也可以用代码下载。
proj = "DHA"
2.生存信息与临床信息
这里仅仅是查看一下,到生存信息部分再整理。
library(GEOquery)
eSet = getGEO("GSE162550",destdir = ".",getGPL = F)
eSet = eSet[[1]]
exp = exprs(eSet)
pd = pData(eSet)
3.表达矩阵行名ID转换
dat = data.table::fread("GSE162550_gene_sample_count_with_symbol (3).xls.gz",data.table = F)
k = dat$Symbol!="---";table(k)
dat = dat[k,]
k2 = !duplicated(dat$Symbol);table(k2)
dat = dat[k2,]
exp = dat[,-(1:3)]
rownames(exp) = dat$Symbol
exp = as.matrix(exp)
4.基因过滤
需要过滤一下那些在很多样本里表达量都为0或者表达量很低的基因。过滤标准不唯一。
过滤之前基因数量:
nrow(exp)
常用过滤标准1:
仅去除在所有样本里表达量都为零的基因
exp1 = exp[rowSums(exp)>0,]
nrow(exp1)
常用过滤标准2(推荐):
仅保留在一半以上样本里表达的基因
exp = exp[apply(exp, 1, function(x) sum(x > 0) >= 0.5*ncol(exp)), ]
nrow(exp)
5.分组信息获取
根据样本ID的第14-15位,给样本分组(tumor和normal)
Group = rep(c("DMSO","DHA"),each = 3)
Group = factor(Group,levels = c("DMSO","DHA"))
table(Group)
6.保存数据
TCGA以外的数据没有clinical,surv,从下面代码里去掉。
save(exp,Group,proj,file = paste0(proj,".Rdata"))
7.三大R包差异分析
rm(list = ls())
load("DHA.Rdata")
table(Group)
#deseq2----
library(DESeq2)
colData <- data.frame(row.names =colnames(exp),
condition=Group)
if(!file.exists(paste0(proj,"_dd.Rdata"))){
dds <- DESeqDataSetFromMatrix(
countData = exp,
colData = colData,
design = ~ condition)
dds <- DESeq(dds)
save(dds,file = paste0(proj,"_dd.Rdata"))
}
load(file = paste0(proj,"_dd.Rdata"))
class(dds)
res <- results(dds, contrast = c("condition",rev(levels(Group))))
#constrast
c("condition",rev(levels(Group)))
class(res)
DEG1 <- as.data.frame(res)
DEG1 <- DEG1[order(DEG1$pvalue),]
DEG1 = na.omit(DEG1)
head(DEG1)
#添加change列标记基因上调下调
logFC_t = 2
pvalue_t = 0.05
k1 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange < -logFC_t);table(k1)
k2 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange > logFC_t);table(k2)
DEG1$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
table(DEG1$change)
head(DEG1)
#edgeR----
library(edgeR)
dge <- DGEList(counts=exp,group=Group)
dge$samples$lib.size <- colSums(dge$counts)
dge <- calcNormFactors(dge)
design <- model.matrix(~Group)
dge <- estimateGLMCommonDisp(dge, design)
dge <- estimateGLMTrendedDisp(dge, design)
dge <- estimateGLMTagwiseDisp(dge, design)
fit <- glmFit(dge, design)
fit <- glmLRT(fit)
DEG2=topTags(fit, n=Inf)
class(DEG2)
DEG2=as.data.frame(DEG2)
head(DEG2)
k1 = (DEG2$PValue < pvalue_t)&(DEG2$logFC < -logFC_t);table(k1)
k2 = (DEG2$PValue < pvalue_t)&(DEG2$logFC > logFC_t);table(k2)
DEG2$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
head(DEG2)
table(DEG2$change)
###limma----
library(limma)
dge <- edgeR::DGEList(counts=exp)
dge <- edgeR::calcNormFactors(dge)
design <- model.matrix(~Group)
v <- voom(dge,design, normalize="quantile")
design <- model.matrix(~Group)
fit <- lmFit(v, design)
fit= eBayes(fit)
DEG3 = topTable(fit, coef=2, n=Inf)
DEG3 = na.omit(DEG3)
k1 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC < -logFC_t);table(k1)
k2 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC > logFC_t);table(k2)
DEG3$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
table(DEG3$change)
head(DEG3)
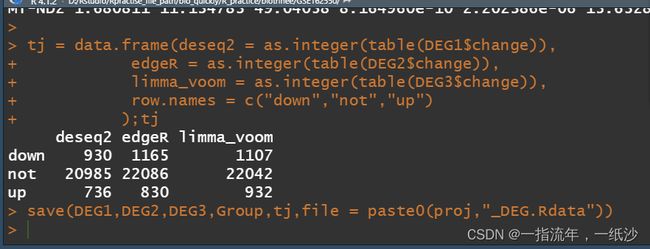
tj = data.frame(deseq2 = as.integer(table(DEG1$change)),
edgeR = as.integer(table(DEG2$change)),
limma_voom = as.integer(table(DEG3$change)),
row.names = c("down","not","up")
);tj
save(DEG1,DEG2,DEG3,Group,tj,file = paste0(proj,"_DEG.Rdata"))
8.可视化
library(ggplot2)
library(tinyarray)
exp[1:4,1:4]
# cpm 去除文库大小的影响
dat = log2(cpm(exp)+1)
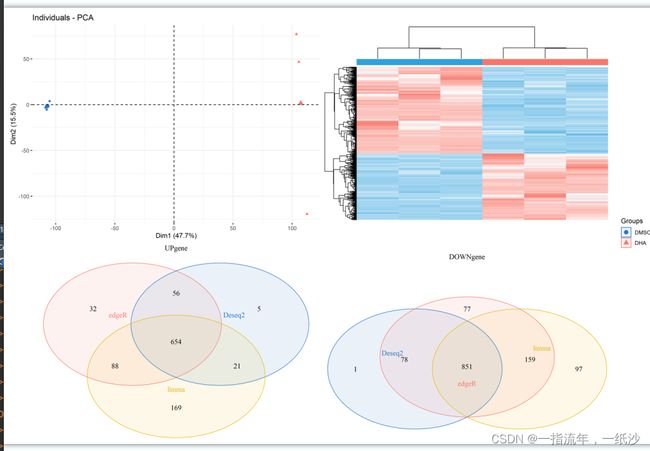
pca.plot = draw_pca(dat,Group);pca.plot
save(pca.plot,file = paste0(proj,"_pcaplot.Rdata"))
cg1 = rownames(DEG1)[DEG1$change !="NOT"]
cg2 = rownames(DEG2)[DEG2$change !="NOT"]
cg3 = rownames(DEG3)[DEG3$change !="NOT"]
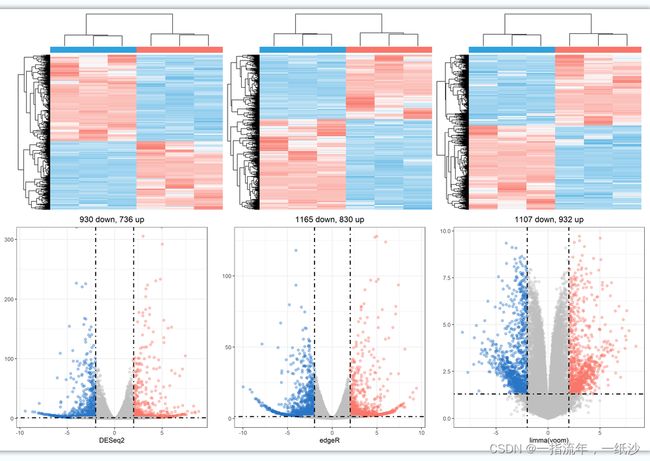
h1 = draw_heatmap(dat[cg1,],Group,n_cutoff = 2)
h2 = draw_heatmap(dat[cg2,],Group,n_cutoff = 2)
h3 = draw_heatmap(dat[cg3,],Group,n_cutoff = 2)
v1 = draw_volcano(DEG1,pkg = 1,logFC_cutoff = logFC_t)
v2 = draw_volcano(DEG2,pkg = 2,logFC_cutoff = logFC_t)
v3 = draw_volcano(DEG3,pkg = 3,logFC_cutoff = logFC_t)
library(patchwork)
(h1 + h2 + h3) / (v1 + v2 + v3) +plot_layout(guides = 'collect') &theme(legend.position = "none")
ggsave(paste0(proj,"_heat_vo.png"),width = 15,height = 10)