使用自监督图像表示学习框架精确预测分子性质和药物靶点(Accurate prediction of molecular properties and drug targets using a sel)
Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework(使用自监督图像表示学习框架精确预测分子性质和药物靶点)
原文链接:Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework
背景
尽管生物医学研究和技术最近取得了进展,药物的发现和开发仍然是一项具有挑战性的多层面任务,需要优化候选化合物的重要特性,包括药代动力学、疗效和安全性。
这一任务的最根本的挑战之一就是如何从化学结构当中去学习分子的表征,当前大部分都是基于特征指纹进行学习的,它需要大量的领域知识。
与传统表征方法相比,自动化分子表征学习模型在大多数药物发现的任务上表现的更好,随着无监督学习在自然语言方面的兴起,SMILES、InChI和二维图也在药物发现任务上得到应用。
本文则鉴于计算机视觉中无监督学习的发展,将其引入到药物发现当中,利用分子的图像作为化合物的特征表示。
模型
模型主要分为三个部分:
- 一个分子编码器,用于从分子图片当中提取潜在特征。(下图中的a)
- 五个预训练模型,通过考虑分子图片中分子的结构特征和它的化学性质等方面,对上一步提取的潜在特征进行调整(下图中的b)。
- 针对下游任务对预训练的分子编码器进行调优(下图中的c)。
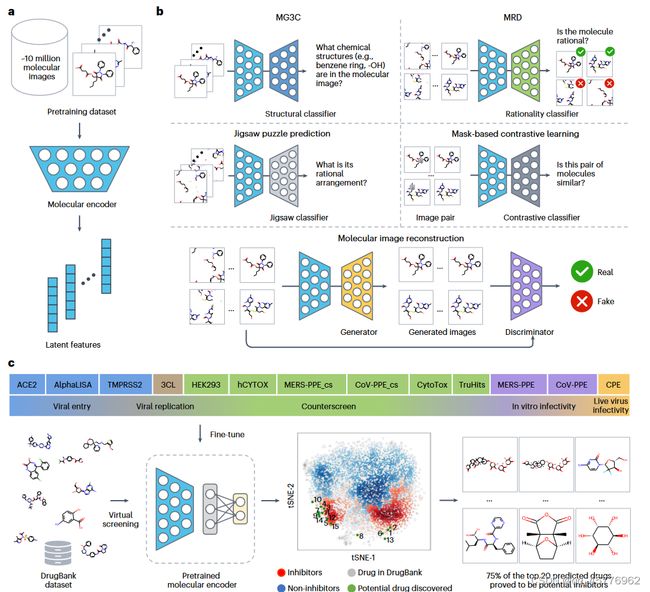
其中第一部分,分子编码器在本文中采用ResNet作为基编码器,通过将分子的图片进行输入,使用该网络的倒数第二层,即最后连接平均池化层的前一层,提取相应的潜在特征。
第二部分是为了根据三大核心原则进行预训练:
- 一致性原则,即相同的化学结构在不同的图片中的语义信息应该是要保持一致的。
- 相关性原则,即对于同一张图片而言不同的图像增强技术在特征空间应当是相关的。
- 合理性原则,即分子结构必须要符合化学的常识。
这三个原则则是通过图b中的五个任务实现。
在一致性原则中,通过使用MACCS密钥(长度166的0-1序列,用于表示分子的结构特征 )进行k均值聚类,在本文中分别使用k值为100,1000,10000进行聚类,对于每个分子获得三个伪标签,使用基于第一部分的基编码器连接三个结构预测线性层,进行预测其伪标签。这就是图b中的MG3C任务,其相应的损失函数为:
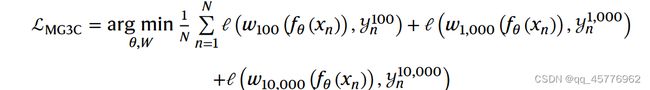
在相关性原则中,通过对在像素级别从潜在特征重建分子图像和最大化掩码图像与原图像之间的关联性进行实现。
在重建分子图像任务中,通过将图像洗牌和重新排列输入到生成模型 G G G中,并期望它能够生成一个正确的图像。这一部分采用的是 G A N GAN GAN 生成对抗网络,其中分子生成器 G G G由4个2D反卷积层和bathNorm层以及ReLu激活函数连结加一个使用tanh激活函数的2D反卷积层组成,而判别器 D D D则由4个2D卷积层和bathNorm层以及ReLu激活函数连结加一个使用sigmod激活函数的2D反卷积层组成,其中分子生成器生成的是64$$64的分子图像,将原图缩放到6464进行比较,这一部分的损失函数为:
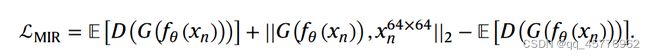
这对应的是图中的Molecular image reconstruction部分。
在最大化掩码图像与原图像之间的关联性任务中采用的是基于掩码的对比学习。对比学习通常需要大量的显式的成对的特征比较,但是这将花费大量的计算机资源,所以本文引入了一种简单的分子图像的对比学习方法,通过将图片划分成16*16的小方格,在这些方格中随机的进行掩码操作,将这一掩码后的图与没有掩码的图同时输入到基编码器当中,查看两者得到的的潜在特征之间的欧几里得距离,作为损失函数:

这一部分对应图b中的Mask-based contrastive learning。
在合理性原则当中,通过重新排列分子图片,进行两个任务,一是判断重新排列后的分子图片是否为正确的分子图片,二是判断重新排列后的分子图片顺序。
这两个任务都是将图片划分为3*3的小块,并将其进行编码和重新排序。
在第一个任务中,通过将图像输入基编码器后的潜在特征,连接一个输出为两维的全连接线性层,并通过一个logic层进行归一化输出。
对应的是图b中的MRD,其相应的损失函数为:
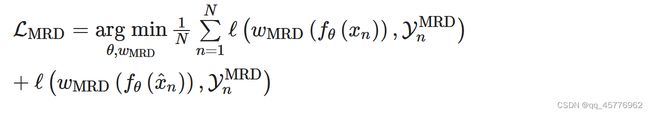
而第二个任务将9*9的图片预先定义101种排列,其中第0个表示原图排列,将图片输入进基编码器,并连接一个101维的全连接层,输出相应的结果,判断是第几种排列。
对应图b重的Jigsaw puzzle prediction,其相应的损失函数为:
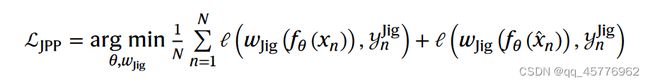
最终反馈给基编码器的损失函数为:

第三部分是下游的模型微调,即针对相应的任务,使用基编码器连接相应的层进行再训练,这一部分并不是文章的重点。主要是通过全连接层连接softmax激活函数获得相应的预测概率,通过交叉熵损失函数进行微调模型。
代码
原作者的代码可能会跑不通,其主要原因在于作者在代码中MRD任务中的损失传递与文中描述的可能有点不一致。
errG = torch.autograd.Variable(torch.Tensor([0.0])).cuda()
errD = torch.autograd.Variable(torch.Tensor([0.0])).cuda()
MIRloss = torch.autograd.Variable(torch.Tensor([0.0])).cuda()
if args.is_recover_training == 1:
real_label = 1
fake_label = 0
################### train D ###################
netD.zero_grad()
label = torch.FloatTensor(data64_non_mask.shape[0]).cuda()
label.data.resize_(data64_non_mask.shape[0]).fill_(real_label)
output = netD(data64_non_mask)
errD_real = criterionBCE(output.flatten(), label)
# errD_real.backward()
# train with fake
hidden_feat_crop, _, _, _, _ = model(Jigsaw_img_var)
fake = netG(hidden_feat_crop)
label.data.fill_(fake_label)
output = netD(fake.detach())
errD_fake = criterionBCE(output.flatten(), label)
# errD_fake.backward()
errD = errD_real + errD_fake
print(errD)
errD.backward()
optimizerD.step()
optimizerD.zero_grad()
################### train G ###################
netG.zero_grad()
label.data.fill_(real_label)
output = netD(fake)
errG_D = criterionBCE(output.flatten(), label)
errG_l2 = (fake - data64_non_mask).pow(2)
errG_l2 = errG_l2.mean()
errG = (errG_D + errG_l2)
MIRloss = (errD + errG) / 2#向外传递的loss
errG.backward()
optimizerG.step()
# optimizer.step()
# optimizer.zero_grad()
optimizerG.zero_grad()
AvgRecoverLoss += (errD.item() + errG.item()) / 2 / len(train_dataloader)
代码中注释的部分即是作者源代码的部分,作者在MIR任务中直接将生成器的loss在该任务过程中直接传递给ImageMol模型的优化器了,从而导致在一轮训练中,该优化器step了两次,从而产生了错误,我选择直接将该部分loss传递到外面总的loss相加。
loss = class_loss * args.cluster_lambda + args.Jigsaw_lambda * Jig_loss \
+ args.constractive_lambda * constractive_loss + \
args.matcher_lambda * reasonability_loss + MIRloss.item()
最后一项就是我修改的部分
结果
本文主要是针对13个SARS-CoV-2靶点的抗病毒的活性进行预测和通过ImageMol识别抗SARS-CoV-2抑制剂这两个任务与其他模型方法进行对比,并再其他公开数据集上进行测试。

模型再各个基准数据集上采用了两种划分数据方式并观察了其相应的多项指标,可以发现模型效果显著,与使用random scaffold split的基于指纹(例如,AttentiveFP11)、基于序列(例如,TF_Robust48)和基于图形(例如,N-GRAM45、GROVER35和MPG37)的模型相比,ImageMol具有更好的性能。
在13个SARS-CoV-2生物检测数据集中,ImageMol获得了从72.6%到83.7%的高AUC值(图3A)。为了测试ImageMol是否能够捕获生物相关特征,我们使用ImageMol的全局平均池层来提取潜在特征,并使用t分布随机邻居嵌入(t-SNE)来可视化潜在特征。图3a显示,ImageMol识别的潜在特征根据它们是所有13个目标或端点上的活跃或非活跃的抗SARS-CoV-2试剂而很好地聚集在一起。这些观察表明,ImageMol可以准确地从分子图像中提取具有区别性的抗病毒特征,用于下游任务。
总而言之,这些综合评估表明,ImageMol在通过不同的病毒靶标和表型分析识别抗SARS-CoV-2分子方面具有很高的准确性。此外,与传统的深度学习预训练模型53或机器学习方法20相比,ImageMol在正负样本极不平衡的数据集上具有更强的能力
通过在ImageMol框架下对3CL蛋白酶抑制剂与非抑制剂数据集的分子图像表示,发现3CL抑制剂和非抑制剂在t-SNE图中被很好地分开(图3B)。活性浓度小于10 μM的分子被定义为抑制剂,否则为非抑制剂。我们显示了DrugBank中每种药物被推断为3CL蛋白酶抑制剂的概率(补充表22),并可视化了它们的总体概率分布(补充图19)。我们发现,前20种药物中有11种(55%)已被确认(包括细胞试验、临床试验或其他证据)为潜在的SARS-CoV-2抑制剂(补充表22),其中两种药物通过生物实验进一步证实为潜在的3CL蛋白水解酶抑制剂。为了测试ImageMol的泛化能力,我们使用了16个实验报告的3CL蛋白酶抑制剂作为外部验证集(补充表23)。ImageMol在16种已知的3CL酶抑制剂中鉴定了10种,并将这10种药物可视化到图3C中的嵌入空间(成功率62.5%,图3D),这表明在抗SARS-CoV-2药物发现中具有很高的泛化能力。