基于张量分解的药物重定位预测药物、靶点和疾病之间的关联
Predicting associations among drugs,targets and diseases by tensor decomposition for drug repositioning
- from BMC Bioinformatics
- 摘要
- 介绍
-
- **药物重定位三类方法**
-
- 基于机器学习
- 基于矩阵分解
- 基于网络
- 基于图谱
- 本文方法
-
- 问题定义
- 数据预处理
- 张量集成
- 药物-蛋白质-疾病关联预测
- 结果
-
- 方法的性能
- 与基线方法比较
-
- 基线方法
- 性能比较
- 新的预测验证
- 潜在的因素分析
- 结论
-
- 恢复丢失的关联
- 影响性能的因素
from BMC Bioinformatics
摘要
药物重定位:发现现有药物的新用途为目标
我们构建并分解包含药物、靶标和疾病之间关联的三维张量,推导出反映这三种实体功能模式的潜在因子。
大多数的预测都是通过文献检索和计算机对接来验证的。利用潜在因素将药物、靶标和疾病归为功能群。
应用拓扑数据分析(TDA)对簇的性质进行了研究。我们发现潜在因素能够捕捉药物、靶标和疾病的功能模式和潜在分子机制。此外,我们专注于重新利用药物治疗癌症,不仅发现新的治疗用途,而且还发现药物的不良影响。
在对药物、靶标和肿瘤亚型之间相关性的深入研究中,我们发现特定簇之间存在很强的相关性。
该方法能够恢复缺失的关联,发现新的预测,并发现药物、靶点和疾病的功能簇。药物、靶标和疾病的聚类,以及聚类间的联系,为药物重新定位提供了新的指导框架。
首先,我们收集药物、蛋白质和疾病之间的成对关联,并将它们整合成一个三维张量,代表药物-蛋白质-疾病三重关联。
然后,对关联张量和一些附加信息进行张量分解,分别得到药物、蛋白质和疾病的三个因子矩阵。
最后,我们利用因子矩阵重构关联张量,得到新的三重关联预测。
我们通过文献搜索和计算对接验证我们排名靠前的预测。此外,我们使用因子矩阵对药物、蛋白质和疾病进行了聚类,因子矩阵反映了药物、蛋白质和疾病的功能模式。将我们的聚类与现有的分类/聚类进行比较,我们发现它们之间存在一些一致性,因子矩阵确实反映了功能模式。
介绍
药物发现是发现潜在新药的过程。这是药学的主要目标之一。传统的实验方法是在一个基因,一种药物,一种疾病的范式下,针对单一靶点设计精巧的选择性配体。然而,它的过程非常耗时和昂贵,并受到高流失率的困扰。此外,这种范式在许多情况下可能会失败:许多药物作用于多个靶点和疾病,而不是在药物设计阶段只作用于一个。不是最初计划的目标被称为脱靶。这种脱靶的相互作用虽然可能引起药物不良反应(ADRs),但为在药物发现中寻求现有药物的新用途提供了机会。
但是,这些年来,成本仍在不断增加,而美国食品药品管理局(FDA)每年用于开发的每一美元所批准的药物数量正在下降。治疗需求和可用药物数量有限之间的差距是药物研究的一大挑战。
基本策略是要么为现有药物找到新的靶点,要么发现新的药物疾病关联。
基本假设是具有相似特性的药物具有相似的治疗效果。
许多药物作用于多个靶点和疾病,而不是在药物设计阶段只作用于一个。最初没有预定的目标称为“脱靶”。这种脱靶的相互作用可能会引起药物不良反应(ADRs),可以为药物发现中寻求现有药物的新用途提供机会。
为了解决关联张量稀疏的问题,我们采用额外的信息来支持我们的关联张量的分解,因为它可以提供更多的关于三联体的信息。
**在本研究中,**我们提出了一种新的基于张量分解的计算药物重新定位方法,不仅为了发现新的药物-蛋白质-疾病三重关联,而且揭示潜在的功能模式。首先,我们从公共数据库中收集药物、蛋白质、疾病及其相应的药物-蛋白质相互作用、药物适应症和蛋白质-疾病关联,并将两两关联/相互作用整合为三维关联张量。其次,我们将整合的关联张量与额外的信息一起进行张量分解,如药物与蛋白质的相似性、药物与药物的相互作用等(DDI)和蛋白质-蛋白质相互作用(PPI)。关联张量分解为三个低秩因子矩阵,表示三重关联的潜在因子以及药物、蛋白质和疾病在潜在因子上的负荷。最后,利用因子矩阵重构原始关联张量,给出新的三重关联。我们将我们的方法与几个基线进行比较,并验证我们的一些新预测。此外,潜在因子负荷用于药物、蛋白质和疾病的聚类,揭示它们的功能模式。
药物重定位三类方法
对接模拟和基于配体的方法是药物重新定位的两种常用计算方法。对接模拟方法[8]利用靶标的结构信息来预测潜在的药物-靶标相互作用,耗时且面临结构信息缺乏的问题。基于配体的方法[9]比较候选配体和目标蛋白的已知配体,以发现潜在的药物-靶标相互作用。然而,对于只有少量相互作用的配体的靶标,基于配体的方法效果不佳。近年来的研究表明,机器学习方法比这两种传统方法更有效,效率更高。
药物-靶标相互作用预测问题可以建模为二分类问题,以药物-靶标对为样本,以药物的化学结构和靶标的氨基酸序列为特征。不同的核被用来建立分类器,如药物和蛋白质的相似性[10]和高斯相互作用谱(GIP)核[11]。为了解决稀疏性和冷启动问题,一些研究采用了半监督学习[12]和邻居信息[1]、[13]。
深度学习技术在图像处理[14]、自然语言处理[15]和语音识别[16]等领域显示出强大的预测能力。近年来也被应用到这一领域。各种深度学习模型已经被使用,如多层感知器[17],[18],深度信念网络[19],堆叠自动编码器[20],[21]。然而,深度学习技术在药物重新定位中的应用仍存在一定的局限性。(1)在训练阶段很难选择阴性样本,因为很少有实验验证的阴性样本。(2)多数研究采用实证特征来代表蛋白质和药物,没有充分利用深度学习的自动学习能力。在传统的机器学习和深度学习方法中,通常很难解释模型的生物学意义。
药物-靶标相互作用预测问题也可以被建模为为给定药物选择潜在蛋白质靶标的推荐问题。矩阵分解是推荐系统中常用的方法,已经以不同的形式应用于药物重新定位,如贝叶斯矩阵分解[22]、概率矩阵分解[2]、逻辑矩阵分解[23]、集体矩阵分解[24]、[25]。与基质类似,药物、蛋白质和疾病之间的联系/相互作用也可以表现为网络。基于网络的推理方法已被用于构建这样的网络,并推断新的关联/交互。一些额外的信息,如药物和蛋白质的结构相似性[4],[26]和副作用信息[27],被整合到网络中,以更准确地预测关联/相互作用。Zheng等人[24]将多种相似度组合成一种方法,证明了不同的相似度在不同的数据集上有不同的性能。
基于机器学习
每一种药物、靶标和疾病都由一个基于它们的性质的特征向量表示,如药物的化学结构、副作用和指纹、靶标的基因组特征、疾病的表型信息等。然后对特征向量进行机器学习模型的训练,进一步提供新的联想预测。
各种深度学习模型被应用于药物再定位,包括多层感知器、深度信念网络堆叠自动编码器等。
在训练机器学习模型时,既要提供正训练样本,也要提供负训练样本。然而,由于该领域很少有实验验证的负样本,因此负样本的选取较为困难。
为了解决稀疏性和冷启动问题,一些研究采用了半监督学习[12]和邻居信息[1]、[13]。
基于矩阵分解
药物重新定位类似于推荐系统,因为它的目的是推荐潜在的治疗疾病的药物。不同种类的信息被用来衡量药物、靶点和疾病的相似性,包括化学结构、遗传变异和基因表达谱。
结果表明,相同的相似度计算方法在不同数据集上的表现不同,而不同的相似度计算方法在同一数据集上的表现也不同。
基于网络
基于图谱
使用特征反转技术来找到具有抗相关表达谱的药物疾病对,基于图谱的方法不需要预先了解疾病或药物的相关性。
然而,当药物或疾病对基因表达没有表现出强烈的干扰时,干扰谱可能导致更高的假阳性。此外,当观察到的变化是疾病的结果而不是原因时,这些方法也可能失败。
miRNA作药物的治疗靶点
本文方法
在这篇文章中,我们提出了一个新的框架来研究药物-靶标-疾病(DTD)三元关联。首先,我们构造代表DTD关联的三维张量,并分解这些张量来推导潜在因素并发现新的预测。我们调查了与药物和靶点相关的不同额外信息的作用以及其他因素的影响。然后,我们检验了预测新关联的能力提出的方法。从关联张量衍生的潜在因素进行分析,以揭示药物、靶点和疾病的功能模式。最后,我们将提出的方法应用于一个癌症数据集,并识别几种癌症的候选药物。
问题定义
在本文中,我们的目标是在已知的两两关系下预测新的药物-蛋白-疾病三重关联,即药物-蛋白相互作用、药物-疾病关联和蛋白疾病关联。为了提高性能,可以使用不同种类的附加信息,包括药物和蛋白质的相似性,以及DDIs和PPIs。因此,输入数据是已知的药物、蛋白质和疾病之间的成对关联/相互作用(图1左),以及一些关于它们的附加信息(图1中),输出是药物-蛋白质-疾病三联体关联的新发现(图1右)。

数据预处理
数据集来自 Luo et al. [28].
在他们的数据集中,从DrugBank数据库中提取了708种药物人类蛋白质参考数据库,1,512个蛋白质(HPRD)和5 603种疾病的比较毒理基因组学数据库。他们从DrugBank中收集相应的药物靶标相互作用和药物-药物相互作用,从HPRD中收集蛋白-蛋白相互作用,从CTD中收集药物-疾病和蛋白-疾病关联。我们去除没有药物-蛋白质相互作用的药物和蛋白质,以及药物-疾病关联小于100或300的疾病,因为与每种疾病相关的蛋白比药物多得多。还剩下549种药物,424种蛋白质和340种疾病。我们提取了相应的1 923种药物-蛋白相互作用、73 075种药物-疾病相互作用、129 563种蛋白疾病相互作用、6 078种药物-药物相互作用和1 029种蛋白-蛋白质相互作用。
药物靶标相互作用预测中最常用的附加信息是药物和蛋白质的相似性。在本文中,药物的相似度是其化学结构产物图的谷本系数。根据蛋白质的氨基酸序列,使用Smith-Waterman评分来计算蛋白质的相似性。
张量集成
为了模拟药物、蛋白质和疾病的三重关联,我们使用三阶张量χ, χijk表示药物i、蛋白j与疾病k的三重关联。当χijk = 1时,可见药物i、蛋白j与疾病k的三重关联;否则,三重关联不存在或仍然未被观察到。
由于大多数相关数据库只提供药物、蛋白质和疾病之间的两两关联/相互作用,我们通过整合这些两两关联/相互作用来构建关联张量。直观地,当且仅当三个条件都满足时,χijk = 1:(1)药物i与蛋白j相互作用,(2)蛋白j与疾病k相关,(3)药物i与疾病k相关,否则,χijk = 0。用这种方法构造的张量用χtri表示。
然而,上述张量集成策略的结果是一个非常稀疏的关联张量(∼0.33%)。为了生成一个更密集的张量并提高预测性能,我们采用了一些研究[32]中使用的假设:如果药物A与蛋白质B相互作用并且蛋白质B与疾病C有关,那么我们可以推断出药物A与疾病C有关。因此提出另一个张量集成策略,χijk = 1:(1)药物i与蛋白j相互作用(2)蛋白j与疾病k相关;否则,χjk = 0。我们得到了另一个关联张量χbi。在χbi中观察到或推断出的三联体关联共598 804条(约7.6%)。
药物-蛋白质-疾病关联预测
我们将药物-蛋白质-疾病三重关联预测问题建模为带有附加信息的张量分解:

其中C、P、D分别为药物、蛋白质和疾病的因子矩阵。M是所使用的附加信息矩阵的个数,这里应该是5。Si是被分解为Ai, di, Bi的附加信息矩阵之一。在我们的例子中,Ai和Bi都是χ的因子矩阵。di用于捕获不同数据源之间的尺度差异。公式的第一部分旨在利用因子矩阵近似原始关联张量。第二部分试图近似补充的信息。第三部分是正则化部分,使低秩矩阵尽可能小。ωmain、ωi和ωreg是不同部分的权重,说明了它们在优化中的重要性。
因子矩阵C、P和D共享长度为R(潜在因子数)的潜在因子维数。三重张量的分解和附加信息矩阵在相应维度上共享相同的因子矩阵。通过将关联张量与附加信息矩阵共同分解,药物,蛋白质和疾病的因子矩阵不仅来自于三重关联,还来自于附加信息,将不同的信息源融合在一起。
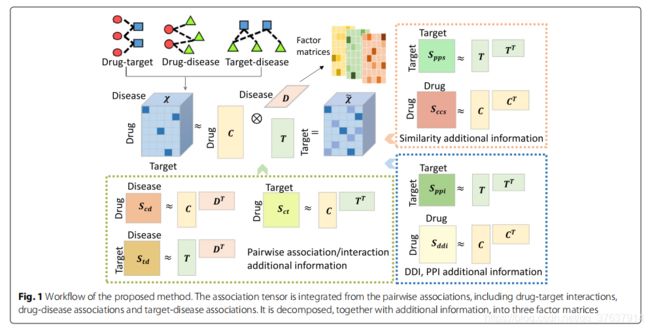
首先我们使用成对关联/相互作用来集成关联张量χ。然后将不同的附加信息对张量进行分解,得到在重构张量χ~的新预测。最后,我们分析了因子矩阵C, P和D。
在这篇论文中,我们使用了不同的附加信息,包括药物和蛋白质的相似性,成对的关联/相互作用,以及DDIs和PPIs(图2底部和右边)。使用成对关联/交互的原因是在张量集成阶段丢失了一些信息,我们希望使用成对关联/交互作为额外的信息来弥补。使用不同附加信息张量分解的详细模型如下:
1)药物-药物相似性和蛋白质-蛋白质相似性

2)成对关联/相互作用

3)药物-药物和蛋白质-蛋白质相互作用

其中Sccs和spp分别是药物和蛋白质的相似性。Scp、Scd和Spd分别是药物-蛋白相互作用、药物-疾病关联和蛋白-疾病关联。Scci为DDIs, Sppi为PPIs。对关联张量进行分解后,由三个潜在矩阵C、P、D重构得到一个新的张量χ,并从χ(图2中)得到新的预测。我们使用Tensorlab包 [33]进行张量分解。
结果
方法的性能
我们使用10倍交叉验证来检验三重组关联预测的性能。我们用受检者工作特性曲线下面积(AUC)和精确召回曲线下面积(AUPR)来评价其性能。成对的关联/交互包含与测试数据相关的信息。因此,当使用它们作为附加信息时,我们从Scp、Scd和Spd中删除测试集中每个三元组关联的所有成对关联/交互。我们将wmain的值设置为1,w1, w2和w3的值设置为5e−3,wg的值设置为1e−6,这是Tensorlab的默认设置。
我们首先研究了性能与潜在因素数量(R的值)之间的关系。在图3中,当使用相似性作为附加信息对关联张量χbi进行分解时,我们显示了AUC和AUPR作为R的函数。我们发现AUC和AUPR随着R的增大而增大,然后在R接近250时收敛。之后性能会下降,因为它过于适应观察到的数据,而不是提取功能模式。在使用其他附加信息时,以及在分解χtri张量时,也会出现类似的趋势。
然后我们比较了用不同的策略,即χtri和χbi来分解张量的性能(图4)。我们发现,对χtri和χbi进行分解的AUC具有可比性,而对χtri进行分解的AUPR却远低于χbi。正如我们所知,在这样一个负样本占很大比例的不平衡预测问题中,即,未观察到的相关或非相关药物蛋白疾病三联体,AUPR被认为比AUC信息更丰富,因为AUC可能给出算法过于乐观的图像,有很多假阳性,因此精度较低。对χtri的分解给出了更多的假阳性预测,特别是在使用相似度作为附加信息时。这可能是由于χtri的稀疏性和相似测度所涉及的噪声所致。
我们进一步研究了在χtri分解中使用不同的附加信息的效果,包括相似度、成对关联/交互、DDIs和PPIs(图5)。我们发现附加信息对AUC的贡献大于AUPR。当R的值足够大时,使用不同信息或不使用额外信息的性能变得可以比较。但是,使用更大的R需要更多的计算资源。使用相似度作为附加信息效果最好,在R较小的情况下,AUC可以提高2%左右。它支持相似的药物可能与相似的蛋白靶点相互作用,这在社会上被广泛接受,并被用作药物-蛋白相互作用预测的基础假设。由于在使用测试样本作为附加信息时,我们删除了所有与测试样本相关的成对关联/交互,因此几乎没有附加信息留下来,以获得比不使用附加信息更好的性能。DDIs和PPIs似乎涉及一些有用但间接的信息,这些信息在药物-蛋白质-疾病关联预测方面不如相似度那么强大。
与基线方法比较
基线方法
我们将我们的方法与SNScore和另外两种基于异构网络的随机游动和集合矩阵分解的基线预测方法进行了比较。
SNScore: SNScore算法根据连接两个节点的中间节点数量和连接权重计算连接概率。在他们的报告表现中,药物-蛋白相互作用、药物-疾病关联和蛋白-疾病关联预测的AUC分别为0.937、0.868和0.871。由于只提供培训平台,他们的数据涵盖了我们所有的数据,所以在评估时,我们随机选择在χbi中输入10%,将这些三重关联投影成成对关联/交互,并检索它们的SNScore。三联体关联的概率由(5)计算,其中Pdrug-pro和Ppro-dis分别为对应的药物-蛋白相互作用和蛋白-疾病关联的概率。AUC和AUPR是基于三联体关联的推断概率Pdrug-pro-dis计算的。
![]()
在异构网络上的随机游走: Chen等人[35]提出了一种药物-靶标相互作用预测算法,称为在异构网络上带重启的基于网络的随机游走(NRWRH)。我们将该方法推广到药物、蛋白质和疾病三种节点的网络,构建出如图6a所示的异构网络。节点的扩散状态被用作成对关联/交互的概率。采用网格搜索的方法求解NRWRH中三个重要参数的最优值,即最大迭代次数、重启概率和转移概率。最后,我们将它们的值分别设置为400、0.5和0.2。在评估时,由于χbi中的关联数远远大于网络中的关联数,我们将测试样本的数量设为NRWRH占结对关联/交互的10%。生成检验样本时,在χbi中随机选取Ntest(由(6)项计算)项,将每个三元组关联划分为三个成对的关联/交互,随机删除其中一个,并通过NRWRH计算其概率。三联体关联也由(5)计算。

其中Ndrug、Nprotein、Ndisease分别为药物数、蛋白质数、疾病数。
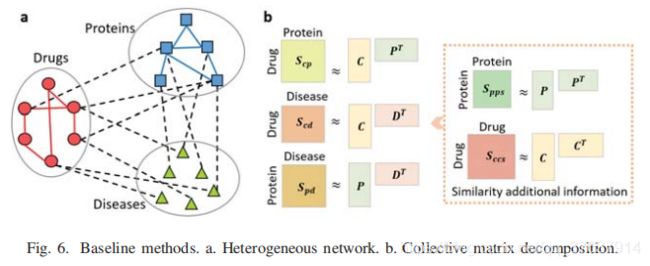
集体矩阵分解: 与我们的方法相似,只是使用了低阶矩阵,我们在图6b中共同分解了5个矩阵。三个成对的关联/交互矩阵作为主矩阵,而两个相似矩阵作为附加信息。我们将潜在因子的数量设置为100,其性能优于200。我们以与NRWRH相同的方式评估这种基线方法。
性能比较
我们将我们的方法与上述三种基线方法进行比较,结果如图7所示,其中RW和MD分别为随机游走法和集体矩阵分解法。在我们的方法中,我们设置R = 250,并使用药物和蛋白质的相似性作为附加信息,这意味着我们使用完全相同的数据RW和MD方法。我们的方法在AUC和AUPR上都优于其他方法。与次优方法RW相比,该方法的AUC和AUPR分别提高了4.07%和40.31%。SNScore对三元组关联的预测效果远低于他们报告的成对交互/关联预测。此外,我们发现尽管RW和MD得到了较高的AUC,但它们得到的aur比我们的方法低得多。
新的预测验证
为了进一步验证我们方法发现新关联的能力,我们调查了50个排名靠前的新预测,即重构张量~χbi中的项具有最大值,而它们在原始j集成张量χbi中的值为零(之前未观测到)。在这一节中,我们使用张量分解的结果R = 250,相似度作为附加信息。由于很难以三联体的方式验证新的预测,我们将它们投射到成对关联/交互中,并试图通过验证这些成对关联/交互来间接验证新的预测。我们检查了排名前50位的三联体预测,并在表I中显示了它们预测的成对关联/相互作用的统计数据。“已知”意味着在输入数据中存在成对的预测关联/交互,而“未知”意味着它们在输入数据中没有被观察到(对应的值为“0”),这是新的成对预测。在前50名三联体预测中,所有相关的药物-蛋白相互作用都是已知的,所有相关的蛋白-疾病关联都是未知的,大部分相关的药物-疾病关联都是未知的。这意味着更容易发现与已知的药物-蛋白相互作用相关的新的三联体。
通过文献搜索,我们在前50名的三重预测中发现了对这些新的成对预测的支持。因为我们输入的数据来源于更新于2013年的CTD,并在2017年发布了新的版本,我们首先在更新版本的CTD中搜索新的成对预测。总共有36种新的药物-疾病联系和50种新的蛋白质-疾病联系。他们在CTD 2017年的推理得分分布如图8所示。0表示我们没有发现2017年CTD中提到的成对关联/交互作用。我们发现超过一半的成对的新预测是支持的,包括一种药物-疾病的关联和47种蛋白质-疾病的关联。他们中的一些人在2017年CTD中获得了较高的推理分数。几乎所有有文献支持的预测都来自蛋白质与疾病的关联。我们用验证过的支持来标记成对的关联/交互。根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。维甲酸受体是核转录调节因子甲状腺激素受体超家族的成员。
根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。它在各种生物过程中调控基因表达。阿维a激活核维甲酸受体,诱导细胞分化,抑制细胞增殖,抑制炎性细胞对组织的浸润。根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。它们可能是新的发现,将被进一步研究。
根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。为了验证我们的方法预测药物蛋白相互作用的能力,我们查看了排名前10000位的预测结果,发现了一些新的药物-蛋白相互作用(表II)。我们选择排名前10位的药物-蛋白相互作用预测结果,在CTD和Pubmed中搜索,找到了其中5个的参考文献。我们使用Autodock Vina[40]对所有排名前10的预测进行计算对接。而有两种蛋白没有3D结构,即CHRM5和PTGER2。我们采用排名第11位的药物蛋白对美替拉酮和FDX1代替PTGER2和比马前列素,没有文献支持。支持参考文献和对接分数见表三。三对氢键的对接位姿和预测氢键如图10所示。
潜在的因素分析
根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。这些因子矩阵通过将原始关联张量和附加信息压缩为低秩矩阵来反映它们的功能模式。因此,通过分析因子矩阵,我们发现功能相似的药物、蛋白质和疾病,并将它们聚到不同的功能组。我们在图11a中展示了药物聚类的结果。我们使用药物的因子矩阵,即D对所有药物进行聚类,并将我们的聚类与DrugBank中药物的化学结构分类进行比较。根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。另一方面,一些不同化学结构类的药物聚在一起,虽然化学结构不同,但功能模式相似。例如,布唑胺和甲唑胺属于有机杂环类化合物,呋塞米属于苯类化合物。这三种药物聚集在一起(图11d)。我们发现它们都具有相同的蛋白质目标碳酸酐酶2。腺苷属于腺苷类或腺苷衍生物,据报道与蛋白靶标腺苷受体A2a相互作用,就像有机杂环类化合物中的另外两种药物茶碱和己酮可可碱一样,它们与腺苷紧密聚集在一起(图11e)。
结论
根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。将我们的方法与一些基准方法进行比较,我们发现我们的方法优于其他方法。此外,我们已经通过文献搜索验证了大多数前50名的新预测。此外,我们还检查了最新预测的10大药物-蛋白相互作用(它们不在前50大预测中)。根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。最后,我们分析了由张量分解得到的因子矩阵,并利用它们对药物进行聚类。通过与现有的药物化学结构类的聚类比较,以及对几种案例的分析,我们发现衍生的潜在因子确实反映了药物的功能模式。
根据我们之前的假设,我们进一步将相应已知或已验证的药物-蛋白相互作用和蛋白-疾病相互作用的药物-疾病关联设置为“部分验证”。我们将与top50预测相关的成对预测在网络中可视化,如图9所示。首先,由于张量分解需要大量的计算资源,目前我们只能研究相对较少的药物、蛋白质和疾病(约500种)。为了进行大规模的关联研究,并行张量分解技术将会有所帮助。第二,除了在药物重新定位中广泛使用的药物和蛋白质的相似性,检查是否使用疾病相似性作为附加信息将有助于提高性能是一项有趣的未来工作。接下来,需要从生物学和病理学的角度对关联和结果进行进一步的研究和解释,以发现一些在现实应用和药物研究中的重要关联。
恢复丢失的关联
关联张量是从两两关联中整合的,包括药物-靶标相互作用、药物-疾病关联和靶标-疾病关联。它与附加信息一起被分解成三个因子矩阵

构建两个关联张量![]()
根据DTD子集使用不同的方法,χtri中的每一项都代表了对应的三重关联的存在,所有这三种成对的关联都存在
每个三重关联在χbi上表示对应的药物,目标和疾病,药物与靶标相关而靶标与疾病相关,因此推断出药物与疾病相关。每个关联张量分别与不同种类的附加信息一起分解,得到药物、靶标和疾病的三个因子矩阵。
我们通过10倍交叉验证测试了该方法的恢复缺失关联的能力,并使用*接收者工作特征曲线下面积(AUC)和精确召回曲线下面积(AUPR)*来评估方法的性能(方法)。
AUC和AUPR随着潜在因素数量的增加而增加。一个可能的原因是,更多的潜在因素具有更高的能力来表征潜在的关联模式,因此它们可以更好地近似张量。然而,当潜在因子的数量进一步增加时,由于导出的潜在因子对关联张量中的观测值过于拟合,因而泛化能力较低,性能下降。
影响性能的因素
我们调查可能影响恢复缺失关联的性能的因素,包括张量构造策略,使用不同种类的附加信息,和张量的稀疏性。